Viewing Molecules¶
sire has integrations with
NGLView and
RDKit to enable you to easily create
two dimensional and three dimensional views of molecules. These
are available via the view2d() and
view() functions that are available for
every molecule, molecule view, collection and system object.
Calling view2d() or
view() will view whatever molecular data
is contained within that object in either 2D or 3D.
2D Views¶
Creating 2D structure views is very straightforward. Simply
call the view2d() member function
of the object that contains the molecular data you want to view.
For example, (in a Jupyter notebook or similar) you can view individual molecules…
>>> import sire as sr
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.top", "ala.crd"))
>>> mol = mols[0]
>>> mol.view2d()
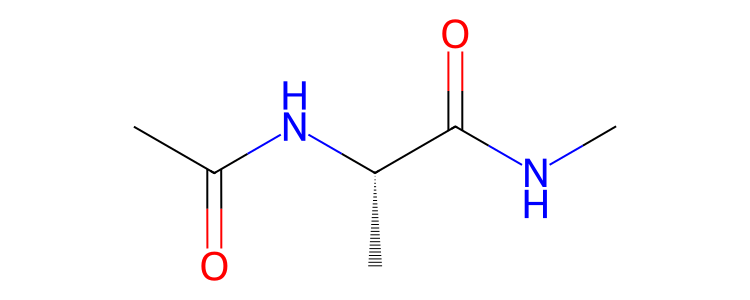
or parts of molecules.
>>> res = mol["residx 1"]
>>> res.view2d()
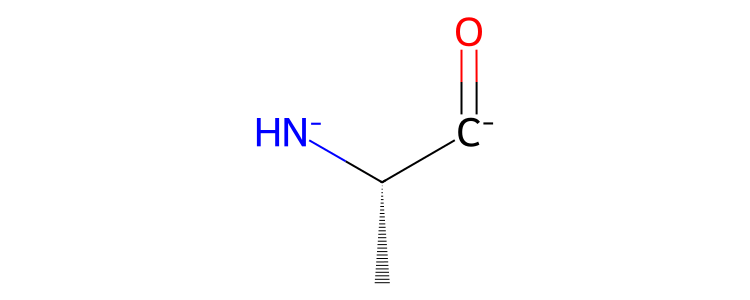
Note
Note that the charge and bond state of partial molecules may be incorrect. This is because the algorithm that assigns charges and bonds will see that the atoms whose bonds have been broken are missing electrons in their valence shell. The algorithm will try to correct this by adding or removing extra bonds, or adding or removing electrons from those atoms.
You can even view collections of molecules. In this case, molecules are grouped together by structure, and you see the number of each type of molecules in the collection.
>>> mols.view2d()
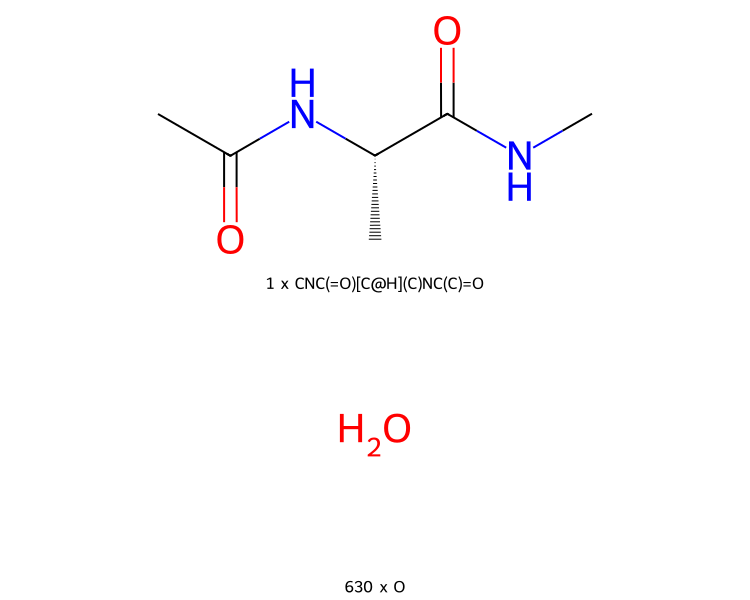
By default, the molecules are printed in a single column. You can
print the molecules across multiple columns by setting their number
via the num_columns argument, e.g.
>>> mols.view2d(num_columns=2)
If you aren’t working in a Jupyter notebook (or similar), or if you want
to save the images to a file, simply pass your desired filename as the
filename argument, e.g.
>>> mols.view2d(filename="structure.png")
/path/to/structure.png'
This returns the full path to the image that was created. The image format
will be chosen based on the file extension. Supported formats are
SVG (.svg), PNG (.png) and PDF (.pdf). Note that you may need
to install the cairosvg library to save to PNG or PDF. If you don’t
have this installed, then a warning will be printed and the image will
be saved in SVG format (with the file extension changed to .svg).
By default, the image size for both displaying in a notebook and saving
to a file is 750x300 pixels for single-molecule views, and
750x600 pixels for multi-molecule views. You can control the image size
via the height and width options, e.g.
>>> mols.view2d(filename="structure.png", height=1000, width=1000)
/path/to/structure.png'
Also, by default, this structure view will only include hydrogens where
they are needed to resolve any ambiguities. Unambiguous hydrogens are
not shown. You can view them by passing include_hydrogens==True, e.g.
>>> mol.view2d(include_hydrogens=True)
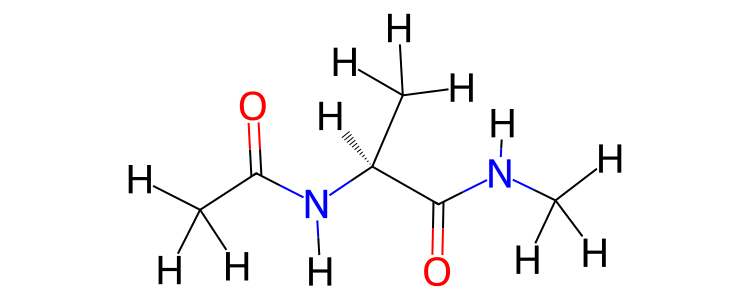
The bond state (single, double, aromatic etc.), formal charge and
stereochemistry of the atoms and bonds in the molecule(s) is determined
automatically if this information is not present within the
molecule(s)’s properties. A simple, yet effective algorithm
described here
has been copied into sire. This algorithm loops over atoms
and adds or removes bonds and electrons such until each atom has filled
its valence shell. The stereochemistry is determined using the
AssignStereochemistryFrom3D
function from RDKit, based on the coordinates in the coordinates
property. As with all of sire, you can change the properties
used to find information from a molecule by passing in a property
map via the map argument of view2d().
3D Views¶
Creating 3D views is similarly straightforward. Simply call
the view() function on the object
that contains the molecule data you want to view. This will start
an interactive 3D viewer that you can use to rotate, translate and
zoom around. If the molecule has multiple trajectory frames, then
you will also get video player controls to play, pause, stop and
scroll through an animation of each frame.
Note
3D views can only be created within Jupyter notebooks (or similar). There is no option currently to let you save the image to a file.
You can view individual molecules…
>>> import sire as sr
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.top", "ala.crd"))
>>> mol = mols[0]
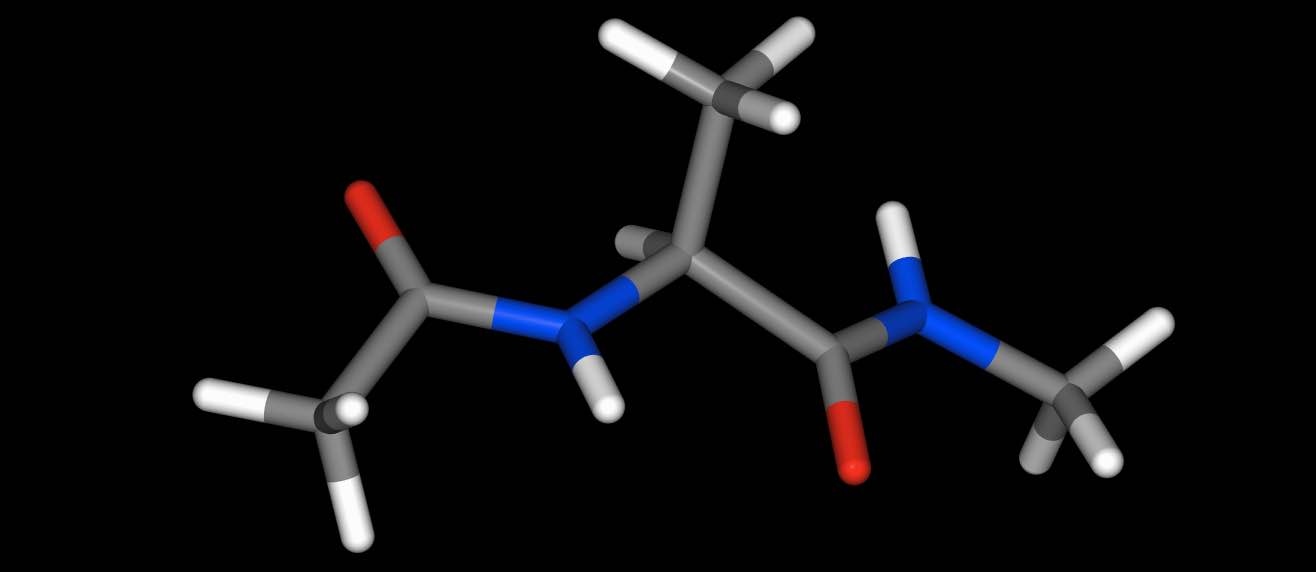
>>> mol.view()
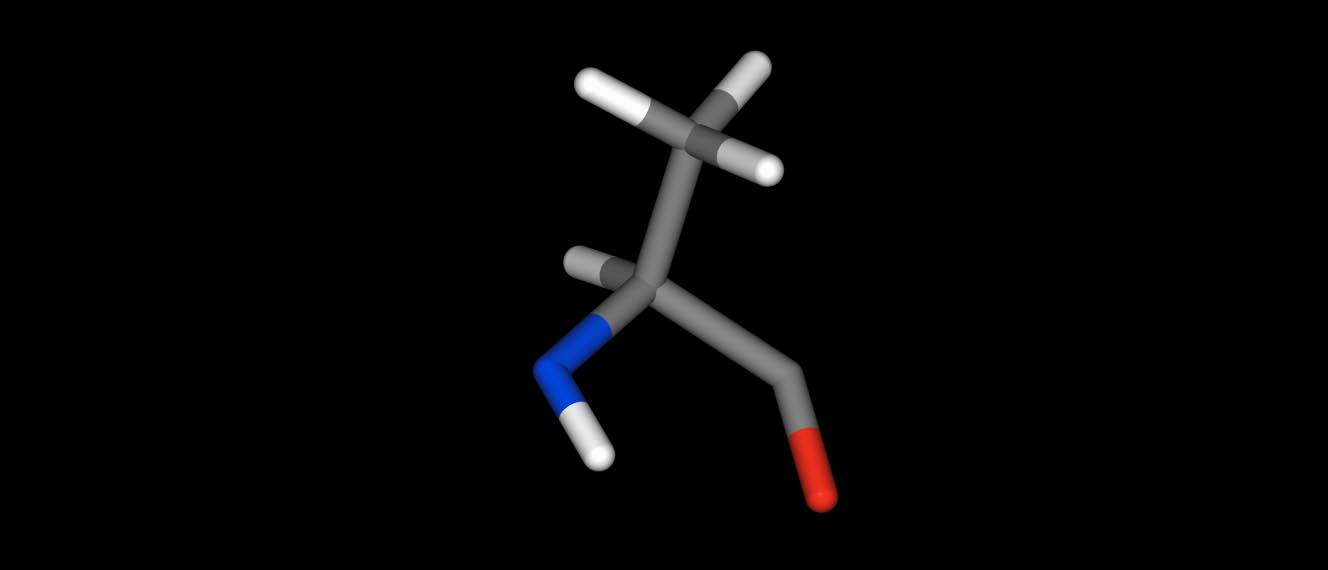
parts of molecules…
>>> res = mol["residx 1"]
>>> res.view()
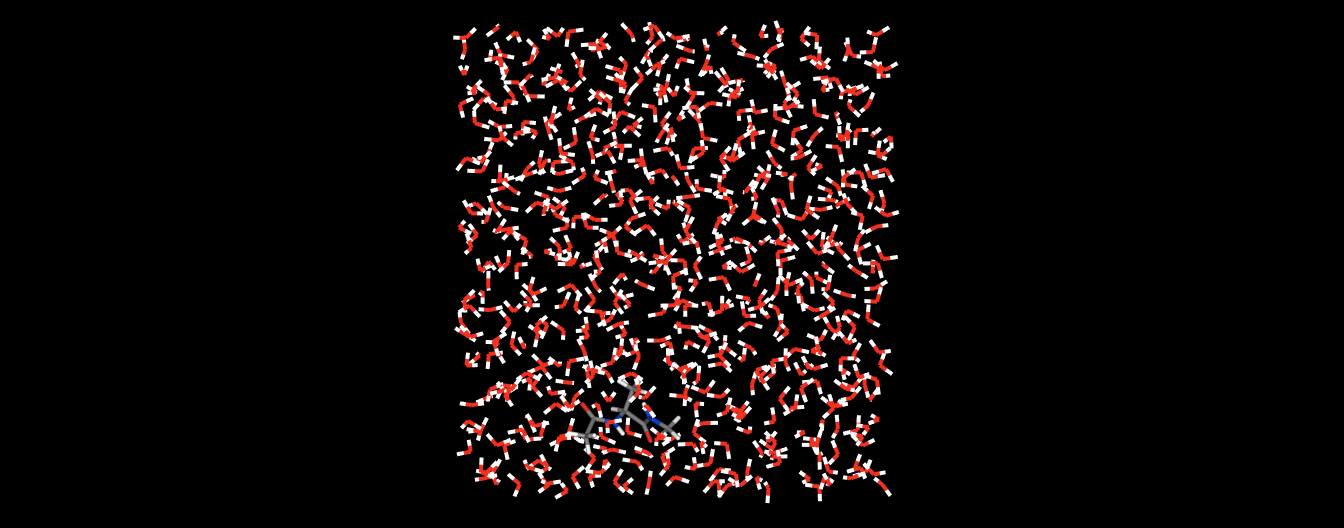
or even whole collections of molecules.
>>> mols.view()
By default, the 3D view is orthographic. You can switch to a perspective
view by passing orthographic=False, e.g.
>>> mol.view(orthographic=False)
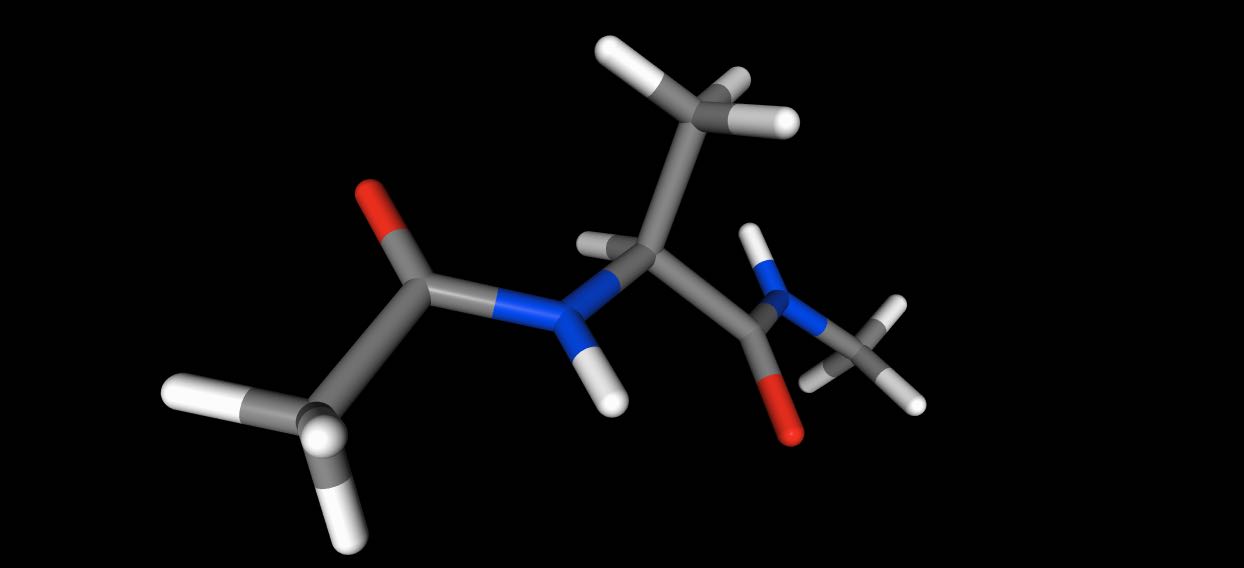
Choosing the 3D representation¶
You can control the representation used for the view via the additional arguments of the function.
protein- set the representation used for protein moleculeswater- set the representation used for water moleculesion- set the representation used for single-atom ionsdefaultorrest- set the representation used for all other molecules (e.g. ligands)
You can also force all molecules to use the same representation by
setting the all option.
Setting any of the above to None, False or the string none will
switch off that view. Setting default to None or False, or
setting no_default to True will disable all default views.
NGLView provides several representations that you can use. These are:
ball_and_stick- a ball and stick viewbase- simplified DNA/RNA base viewcartoon- traditional “cartoon” view of a proteinhyperball- smoothly-connected ball and stick viewlicorice- prettier line viewline- simple line viewpoint- simple point for each atomribbon- show the protein backbone as a ribbonrocket- rocket viewrope- show the backbone as a ropespacefill- Spacefilled spheres for each atomsurface- Render the molecular surface onlytrace- trace viewtube- show the backbone as a rope
So setting protein="tube" would render protein molecules with a
tube representation. Setting all="spacefill" would render
all atoms using a spacefill representation etc.
The following default representations will be used:
protein-cartoon:sstrucwater-line:0.5ion-spacefilldefault-hyperball
Note
The sstruc and 0.5 values refer to colors, which are described
in the next section.
Note
We use default to refer to any other molecule, e.g. typically
ligands.
You can switch off the default representations by passing
no_default=True, e.g. mols.view(no_default=True, protein="surface")
would show only the surface view of a protein. You can also switch off
all default representations by passing default=False, default=None,
all=False or all=None.
You can also pass multiple representations per view by passing in a list of representations, e.g.
>>> mols = sr.load("3NSS")
>>> mols.view(protein=["tube", "licorice"])
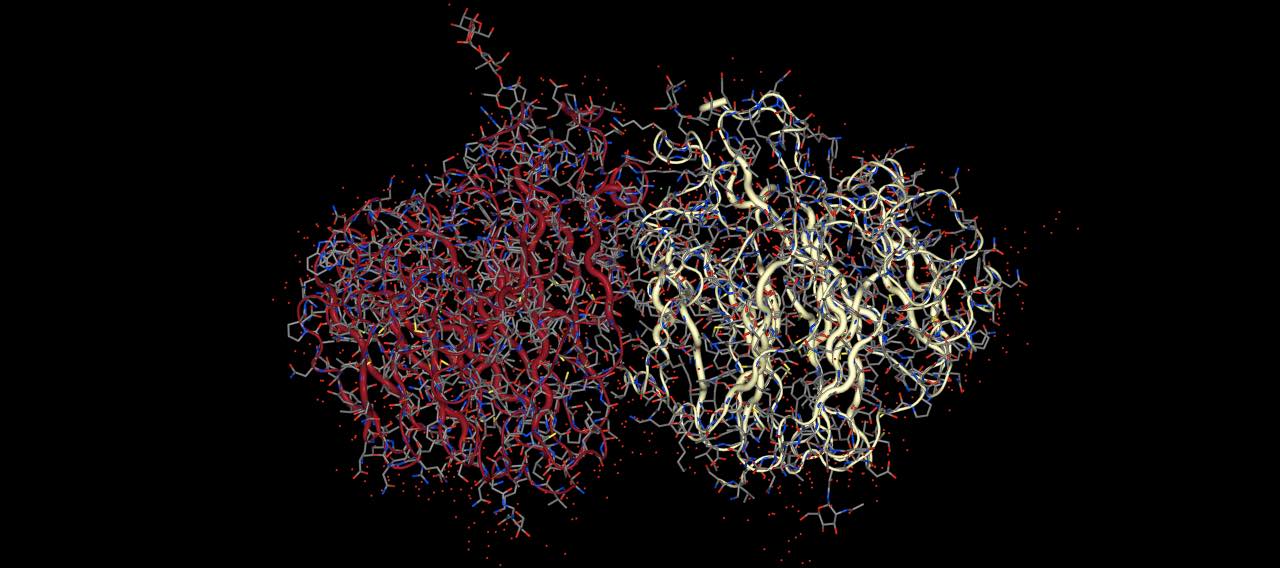
views the protein with both a licorice and a tube representation.
The terms protein, water and ion are performing searches
of the molecule(s) for all atoms that match those search terms.
You can create your own selections by passing in search terms for
arguments that match the representation. For example
>>> mols.view(spacefill="resname ALA")
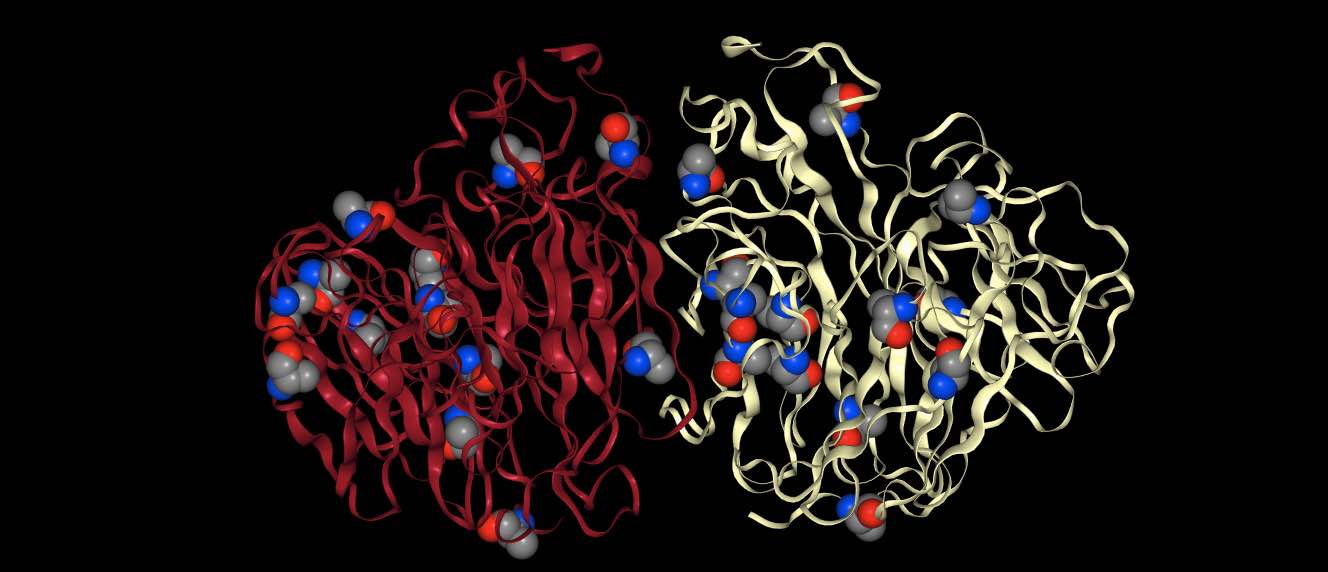
will render the protein in default view (cartoon) and will additionally
render every atom that matches resname ALA in spacefill.
You can match multiple search terms by passing them in as a list, e.g.
mols.view(spacefill=["resname ALA", "resname ASP"]) would render
both ALA and ASP residues in spacefill.
You can use any search term against any of the representations.
For example, here we will do a more complex view of the aladip system
where we render water molecules that are close to aladip differently
to the rest of the water molecules in the box.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.top", "ala.crd"))
>>> mols.view(no_default=True,
... surface="molidx 0",
... spacefill="water within 5 of molidx 0",
... ball_and_stick="water within 10 of molidx 0",
... rest="line")

Note
Note how the distance calculation takes into account the periodic
boundaries of the system. Note also that you can mix representation
based views (e.g. surface="molidx 0") with search based views
(e.g. rest="line").
Choosing colors and opacities¶
You can set the color and opacity used for a particular representation
by passing these as additional terms to the representation or search
term, separated by colons. For example, to set the color of a
representation to blue we could pass this as an addition :blue
to the representation or search term argument.
>>> mol = mols[0]
>>> mol.view(all="licorice:blue")
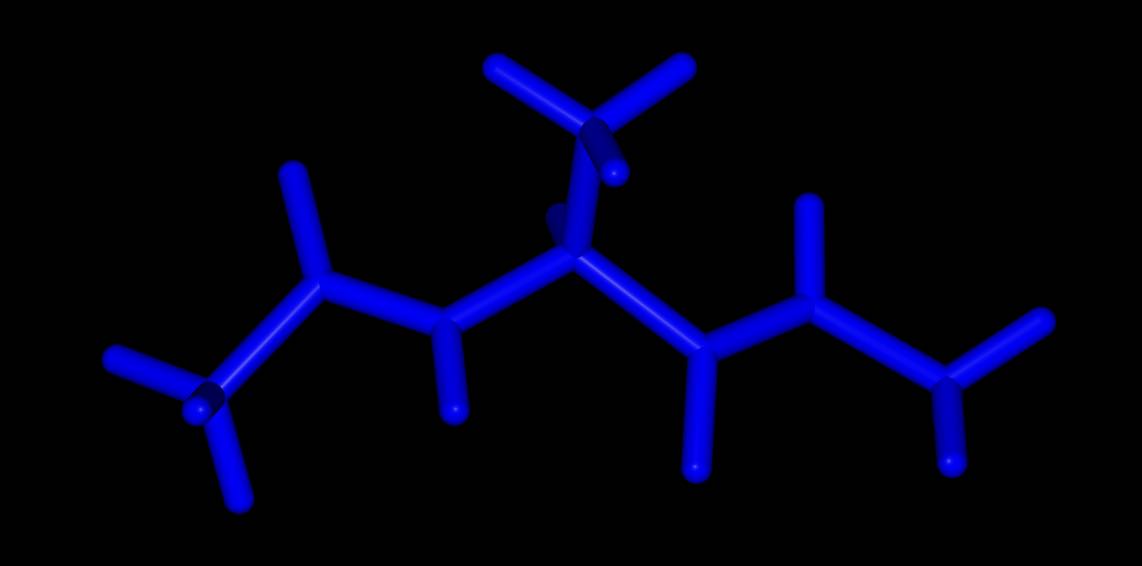
Here all of the atoms are rendered in blue licorice. Or…
>>> mol.view(all="licorice:blue", ball_and_stick="element C:red")
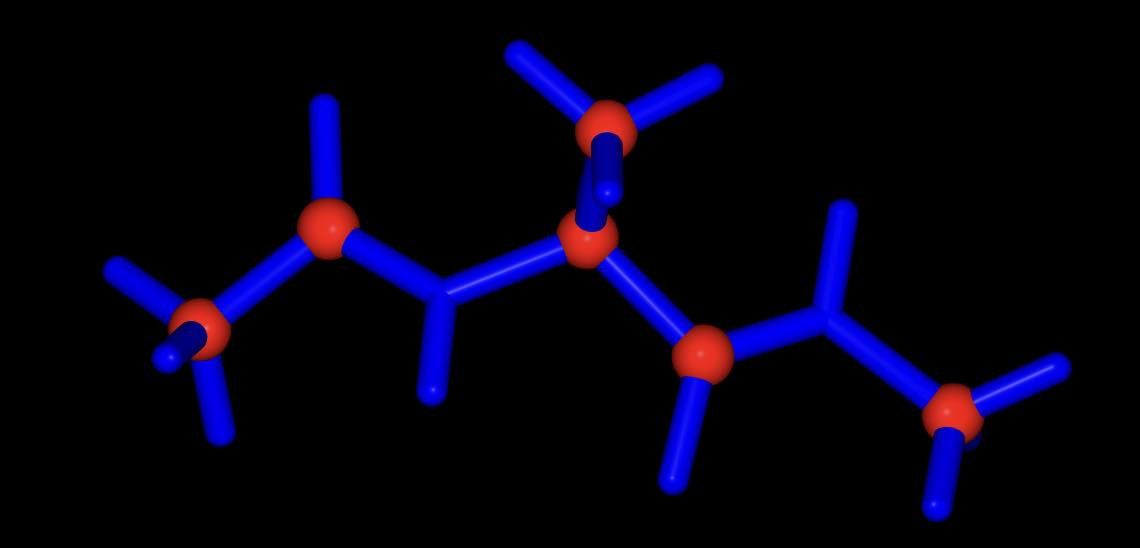
all of the atoms are rendered in blue licorice, but the carbon atoms are represented as red balls and sticks.
You can use any color name supported by NGLView. These include named
colors (e.g. red, green, blue, yellow, including any
CSS named color supported
by your browser, e.g. orchid, sienna, wheat etc.), colors
specified as a red-green-blue hex values (e.g. #FF0000, #00FF00,
#0000FF etc.), colors specified as red-green-blue triples
(e.g. rgb(255,0,0), rgb(0,255,0), rgb(0,0,255) etc.) or any of the
coloring schemes supported by NGLView
(e.g. atomindex, bfactor, electrostatic, element,
hydrophobicity, random or sstruc).
You also specify the opacity (transparency) of the representation by adding a number between 0 (fully transparent) and 1 (fully opaque). You can use any order of color and opacity, e.g.
>>> mol.view(all=["licorice", "spacefill:0.8", "surface:red:0.2"])
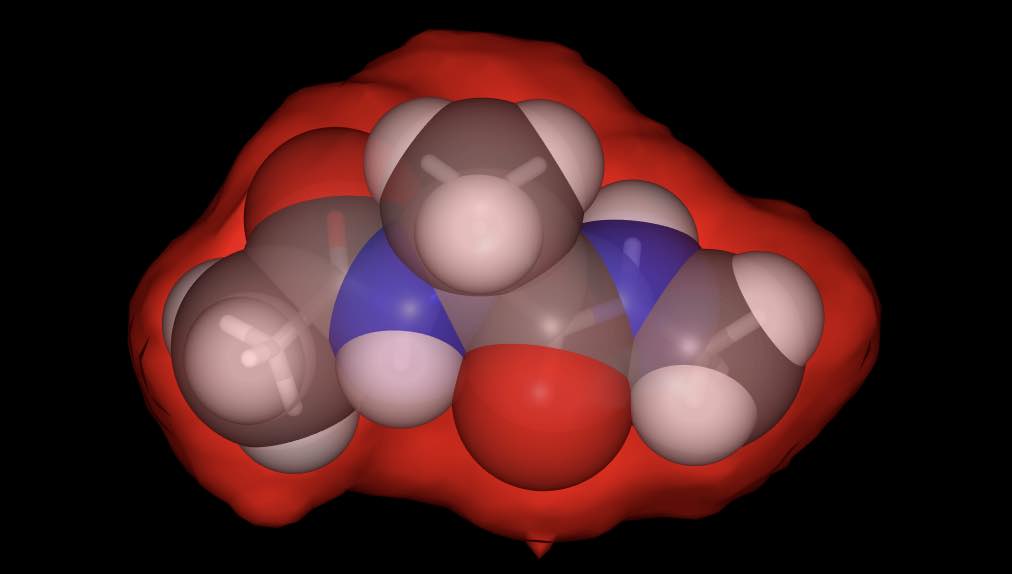
has rendered the molecule using three representations; a licorice in default colors (colored by element), spacefill in default colors, but with opacity 0.8, and a red-colored surface with opacity 0.2.
Or…
>>> mol.view(all=["ball_and_stick", "surface:0.9:electrostatic"])
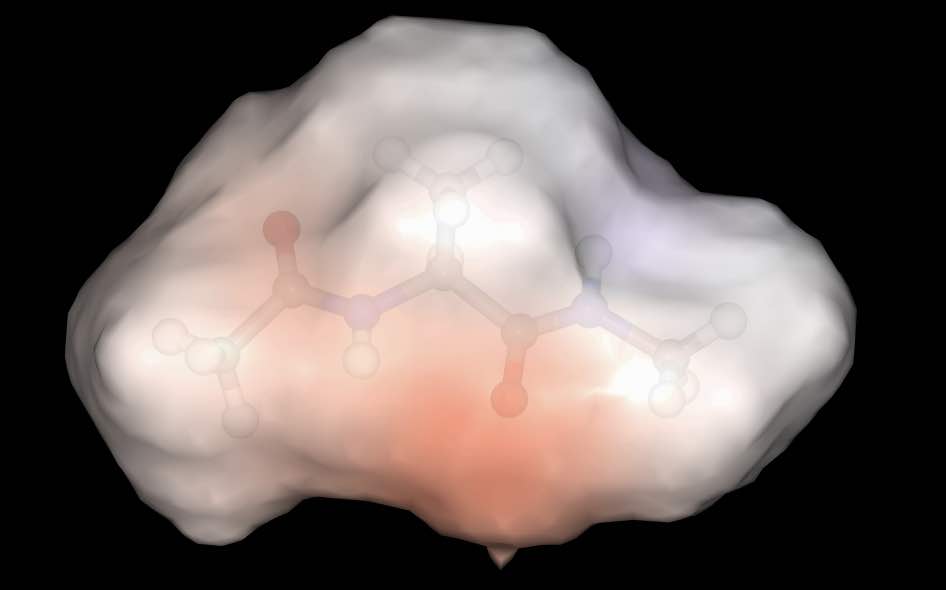
has rendered the molecule with two representations; a ball and stick with default colors and a surface colored using electrostatic potential, with opacity 0.9.
Centering the view¶
You can center the view on any selection using the center option, e.g.
>>> mols.view(center="molidx 0")
would center the view on the first molecule, or,
>>> mols.view(center="not (water or protein")
would center the view on all none (water, protein) molecules, i.e. likely any ligands or ions. Remember that you can create your own custom selections to set search terms that refer to ligands or ions more specifically.
Viewing trajectories¶
If the molecules being viewed have a trajectory, then you will also see play controls in the bottom left. These will let you play, pause and scroll through the trajectory. Click the play button multiple times to speed up playback.
You can choose a subset of frames to play, e.g. here we will play a movie of the first 10 frames of the trajectory.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.top", "ala.traj"))
>>> mols.trajectory()[0:10].view()
Or here we can view every 25 frames of the trajectory.
>>> mols.trajectory()[0::25].view()
Or here we can view all of the frames in reverse order
>>> mols.trajectory()[::-1].view()
Wrapping molecules into the current box¶
You can control whether or not molecules are wrapped into the same
periodic box using the wrap option. If this is True (the default),
then the molecules are wrapped into the same box. If this is False
then the wrapping will be whatever was loaded from the trajectory
file (or generated via the simulation). For example, the trajectory
is not wrapped, and so the water molecules will gradually drift out
into neighboring boxes. You can see this by passing in wrap=False,
e.g.
>>> mols.trajectory()[-1].view(wrap=False)

This compares to
>>> mols.trajectory()[-1].view(wrap=True)
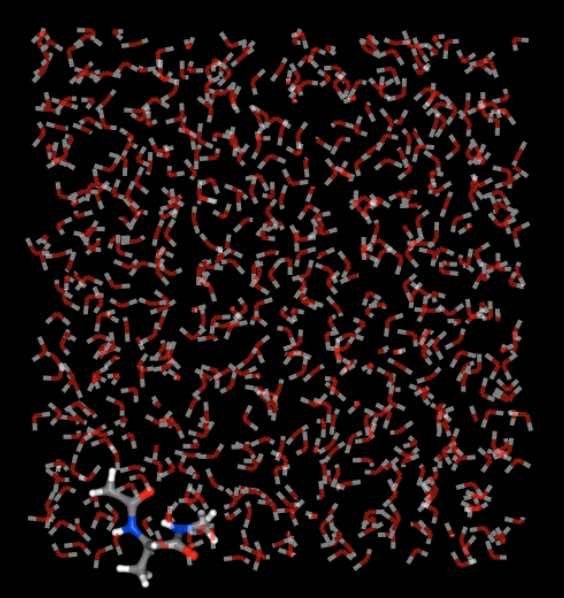
Note
We are viewing the last frame here, as this is the one that shows the maximum amount of drift from the central box.
Trajectory Alignment¶
You can align the frames in a trajectory by passing in a search string
via the align keyword. For example, here we could align every frame
against all of the carbon atoms.
>>> mols.view(align="element C")
Note
You can also pass in a sub-view directly, e.g.
mols.view(align=mols["element C"]).
If wrap is True (as is the default) then all molecules will be wrapped
such that the aligned atoms are at the center of the box.
You can use any search string or view to find the atoms to align. If you pass
align=True then this will align against all atoms. This can be useful
when you are viewing individual molecules, e.g.
>>> mols[0].view(align=True)
Trajectory Smoothing¶
Molecular dynamics trajectories can be difficult to view because there is a
lot of high frequency random motion that can obscure the low frequency
conformational changes that are often of more interest. One way to view
these events is to average the coordinates of atoms over several frames.
You can do this using the smooth option, e.g.
>>> mols.view(align="element C", smooth=50)
Would align the trajectory using all carbon atoms, and would average the coordinates of each atom over 50 neighboring frames.
Note
The smoothing number, s is divided by two, with each frame being
averaged over the previous (s/2)-1 preceeding frames and (s/2)+x
following frames (where x is zero if s is even, and one
otherwise).
Integration with trajectories¶
Note that all of the above options can be passed to the
trajectory() function, e.g.
>>> mols.trajectory(align="element C", smooth=50).view()
would give the same view as
>>> mols.view(align="element C", smooth=50)
as would
>>> mols.trajectory(align=mols["element C"], smooth=50).view()
and
>>> mols.view(align=mols["element C"], smooth=50)
This is because mols.view(...) is creating its own
mols.trajectory(...) with those options, and is viewing those.
While you can do this, and it is useful for, e.g. saving trajectories,
there are lots of edge cases that could trip you up. We recommend that
you pass the options to the view function and don’t pass them to
the trajectory function when you want to view.
Note
The options passed to view will override those passed to
trajectory, e.g. mols.trajectory(smooth=10).view(smooth=50)
will use a smooth value of 50.
Note
The value of wrap in the view function defaults to True.
This means that mols.trajectory(wrap=False).view() will wrap
the coordinates, as the value of wrap in the view function
defaults to True and overrides the value in trajectory.
To show unwrapped, you would need to use
mols.trajectory(wrap=False).view(wrap=False), or the much easier
mols.trajectory().view(wrap=False), or the easiest
mols.view(wrap=False).
Setting the background color¶
You can set the background color of the view using the bgcolor argument.
This should be a string, using any color that is recognised by NGLView
via the backgroundColor stage parameter.
>>> mols[0].view(bgcolor="black")
Note
The default background color is black.
Closer integration with NGLView¶
We don’t yet properly expose the
NGLView stage parameters.
Currently you can pass in a dictionary of parameters via the
stage_parameters argument, which are straight passed to the
NGLView.NGLWidget.stage.
If you want more control over the view, you can assign the result
of mols.view(...) to a variable. This variable is the actual
NGLView.NGLWidget,
which you can manipulate as if you had created it yourself, e.g.
>>> v = mols[0].view()
>>> v.stage.set_parameters(backgroundColor="white")
>>> v.display(gui=True)
>>> v.camera = "perspective"
>>> v
Saving 3D views to files¶
NGLView is not really designed to render frames via a script. Instead, it
needs to be run within a Jupyter notebook so that it has access to the
WebGL view in the web browser to perform the rendering.
This FAQ answer
shows how you could automate rendering images to files. Note that you need
to get the handle to the NGLView.NGLWidget,
as above, and then call render_image to get the image. You will need
to actually view the object in a code cell, and will need to wait for the
image to render, e.g. in one code cell
>>> v = mols.view()
>>> v
Then in the next
>>> def render(view, filename):
... import time
... image = view.render_image()
...
... while not image.value:
... time.sleep(0.1)
...
... with open(filename, "wb") as f:
... f.write(image.value)
and then finally, to call this function in a background thread…
>>> import threading
... thread = threading.Thread(
... target=render,
... args=(v, "image.png")
... )
>>> thread.daemon = True
>>> thread.start()