Loading a molecule¶
We load molecules using the sire.load() function. This accepts either
a filename, a URL, a PDB code or a Uniprot
code to download from the alphafold database.
Loading using a PDB code¶
Let’s first load a molecule from the PDB. You do this by passing the
PDB code as the argument to sire.load(). We will load structure
3NSS;
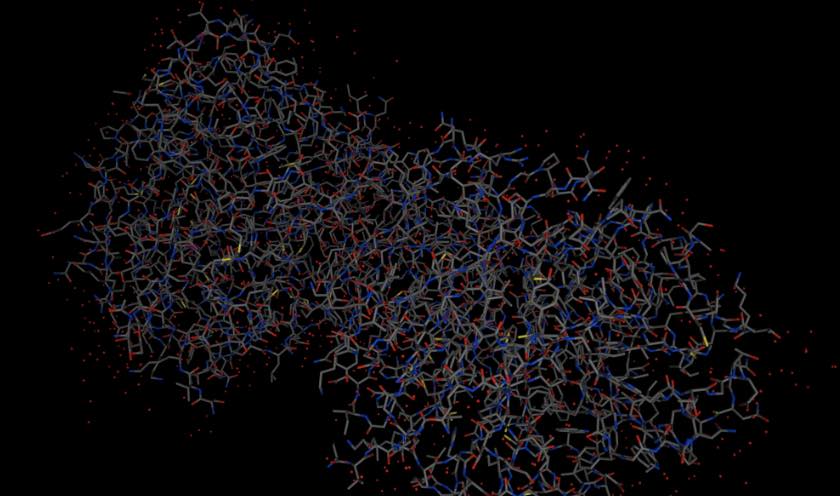
>>> mols = sr.load("3NSS")
>>> print(mols)
Downloading from 'https://files.rcsb.org/download/3NSS.cif.gz'...
Unzipping './3NSS.cif.gz'...
System( name=3NSS num_molecules=1 num_residues=1679 num_atoms=6984 )
Note
This will download a PDBx/mmCIF file from the RCSB website if gemmi is installed. Otherwise, it will download an (older format) PDB file. You can use either traditional (“3NSS”) or new-style (“pdb_00003nss”) PDB codes.
If you are running in a Jupyter Notebook (or similar) you can view
the molecule by calling the view() function,
e.g.
>>> mols.view()
Note
You may see a warning instructing you to install nglview.
If you see this, install nglview by typing
conda install nglview at the command line.
This uses nglviewer to view the molecule.
There are lots of options to the view()
function, which are described in full here.
Loading from Alphafold using a Uniprot code¶
You can also load structures directly from the
alphafold database. To do this,
call sire.load() passing in alphafold: followed by the
Uniprot code. You can search for Uniprot codes via the
search on the alphafold website.
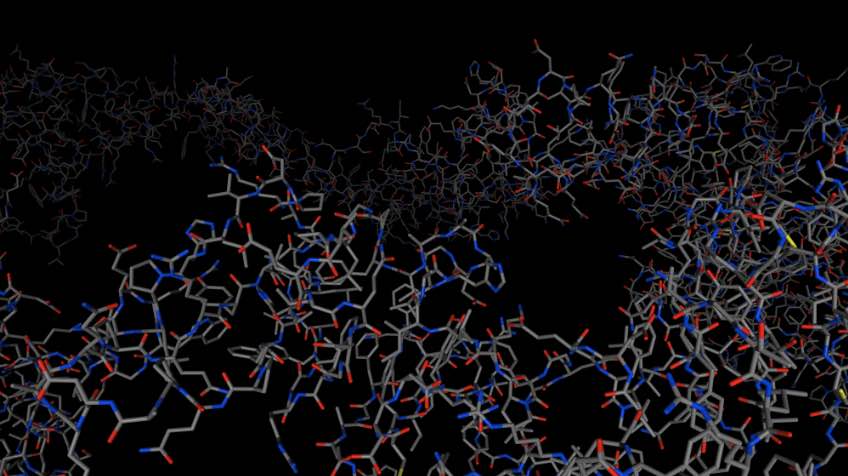
>>> mols = sr.load("alphafold:A0A538R8Y3")
>>> print(mols)
Downloading from 'https://alphafold.ebi.ac.uk/files/AF-A0A538R8Y3-F1-model_v3.pdb'...
System( name=AF-A0A538R8Y3-F1-model_v3 num_molecules=1 num_residues=1190 num_atoms=9417 )
Again, you can use mols.view() to view
the molecules.
>>> mols.view()
Loading from files¶
You can, of course, load molecules directly from files. These can be files that are already downloaded to your computer, or that are available via a URL on the internet. To do this, simply pass in the path to the file on your disk, or the URL of the file on the internet.
For example, let’s load a cholesterol molecule from https://sire.openbiosim.org/m/cholesterol.sdf.
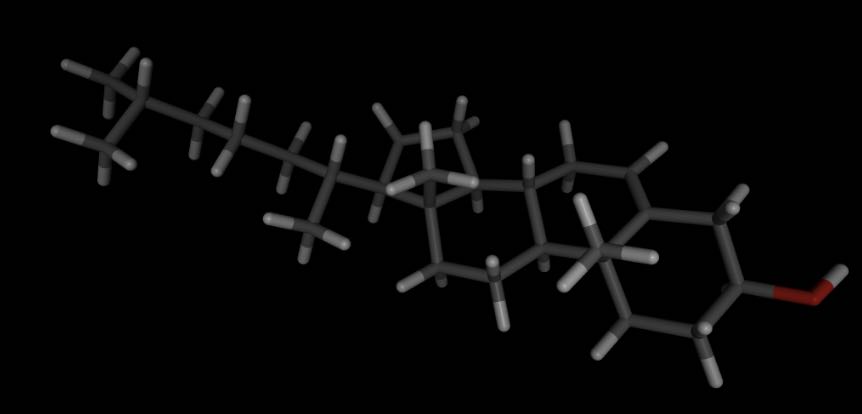
>>> mols = sr.load("https://sire.openbiosim.org/m/cholesterol.sdf")
Downloading from 'https://sire.openbiosim.org/m/cholesterol.sdf'...
Unzipping './cholesterol.sdf.bz2'...
>>> print(mols)
System( name=cholesterol num_molecules=1 num_residues=1 num_atoms=74 )
>>> mols.view()
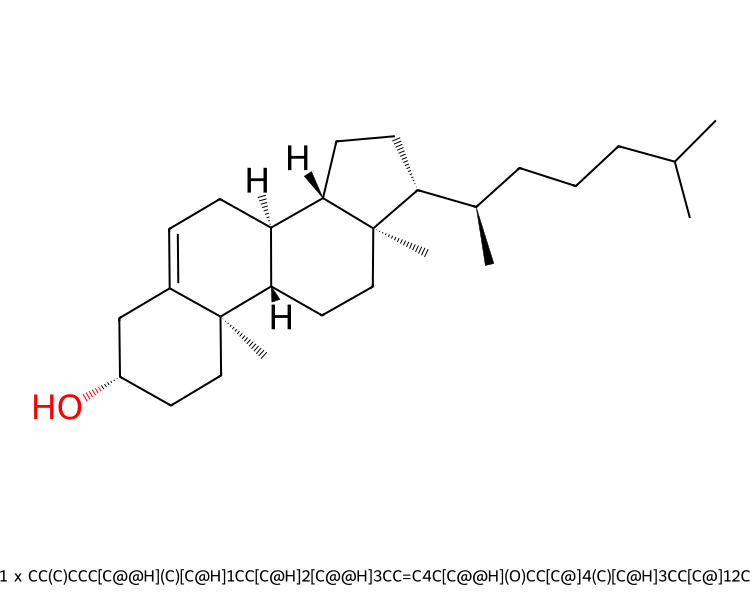
or viewed as a 2D structure…
>>> mols.view2d()
Accessing the molecules¶
Molecules are loaded into a System. You can see how
many molecules have been loaded using the
num_molecules() function;
>>> print(mols.num_molecules())
1
In this case, one molecule has been loaded. You can access this molecule via;
>>> mol = mols[0]
>>> print(mol)
Molecule( MOL:2 num_atoms=74 num_residues=1 )
This shows that the molecule is called MOL and has molecule number 2.
It contains 74 atoms in 1 residue.