Quick Start Guide¶
Import sire using
>>> import sire as sr
Load a molecule from a URL, via sire.load().
>>> mols = sr.load(f"{sr.tutorial_url}/p38.pdb")
Note
sire.tutorial_url expands to the base URL that contains
all tutorial files.
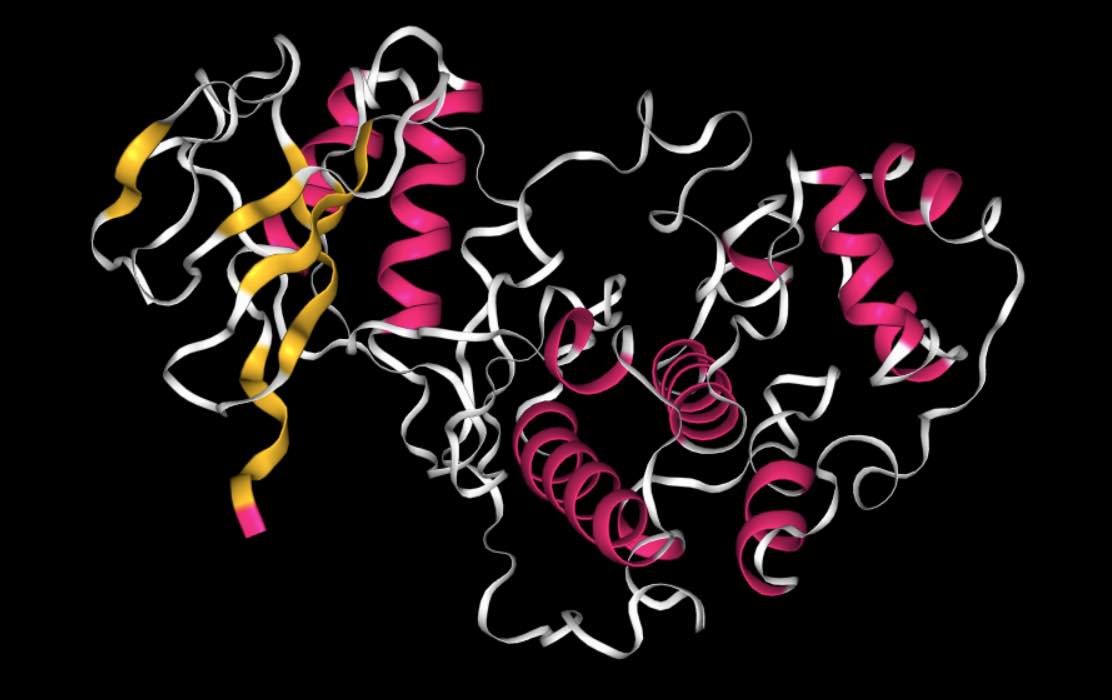
View molecules using view().
>>> mols.view()
Or load molecules that need multiple input files by passing in multiple files.
>>> mols = sr.load(f"{sr.tutorial_url}/ala.top", f"{sr.tutorial_url}/ala.traj")
Note
You could use sire.expand() to put sire.tutorial_url in front
of ala.top and ala.crd, e.g. via
sr.expand(sr.tutorial_url, ["ala.top", "ala.traj"])
There are lots of ways to search or index for molecules, e.g.
>>> mols[0]
Molecule( ACE:7 num_atoms=22 num_residues=3 )
has returned the first molecule in the system of molecules that were loaded.
>>> mols["water"]
SelectorMol( size=630
0: Molecule( WAT:4 num_atoms=3 num_residues=1 )
1: Molecule( WAT:5 num_atoms=3 num_residues=1 )
2: Molecule( WAT:6 num_atoms=3 num_residues=1 )
3: Molecule( WAT:7 num_atoms=3 num_residues=1 )
4: Molecule( WAT:8 num_atoms=3 num_residues=1 )
...
625: Molecule( WAT:629 num_atoms=3 num_residues=1 )
626: Molecule( WAT:630 num_atoms=3 num_residues=1 )
627: Molecule( WAT:631 num_atoms=3 num_residues=1 )
628: Molecule( WAT:632 num_atoms=3 num_residues=1 )
629: Molecule( WAT:633 num_atoms=3 num_residues=1 )
)
has returned all of the water molecules,
while
>>> mols[0]["element C"]
Selector<SireMol::Atom>( size=6
0: Atom( CH3:2 [ 18.98, 3.45, 13.39] )
1: Atom( C:5 [ 18.48, 4.55, 14.35] )
2: Atom( CA:9 [ 16.54, 5.03, 15.81] )
3: Atom( CB:11 [ 16.05, 6.39, 15.26] )
4: Atom( C:15 [ 15.37, 4.19, 16.43] )
5: Atom( CH3:19 [ 13.83, 3.94, 18.35] )
)
has returned all of the carbon atoms in the first molecule.
Smarts searchs are supported too! For example, here are three matches for a smarts string that finds aliphatic carbons.
>>> mols["smarts [#6]!:[#6]"]
AtomMatch( size=3
0: [2] CH3:2,C:5
1: [2] CA:9,CB:11
2: [2] CA:9,C:15
)
You can also search for bonds, e.g.
>>> mols[0].bonds()
SelectorBond( size=21
0: Bond( HH31:1 => CH3:2 )
1: Bond( CH3:2 => HH32:3 )
2: Bond( CH3:2 => HH33:4 )
3: Bond( CH3:2 => C:5 )
4: Bond( C:5 => O:6 )
...
16: Bond( N:17 => H:18 )
17: Bond( N:17 => CH3:19 )
18: Bond( CH3:19 => HH31:20 )
19: Bond( CH3:19 => HH32:21 )
20: Bond( CH3:19 => HH33:22 )
)
has returned all of the bonds in the first molecule, while
>>> mols.bonds("element O", "element H")
SelectorMBond( size=1260
0: MolNum(4) Bond( O:23 => H1:24 )
1: MolNum(4) Bond( O:23 => H2:25 )
2: MolNum(5) Bond( O:26 => H1:27 )
3: MolNum(5) Bond( O:26 => H2:28 )
4: MolNum(6) Bond( O:29 => H1:30 )
...
1255: MolNum(631) Bond( O:1904 => H2:1906 )
1256: MolNum(632) Bond( O:1907 => H1:1908 )
1257: MolNum(632) Bond( O:1907 => H2:1909 )
1258: MolNum(633) Bond( O:1910 => H1:1911 )
1259: MolNum(633) Bond( O:1910 => H2:1912 )
)
has returned all of the oxygen-hydrogen bonds in all molecules.
If a trajectory has been loaded (as is the case here) then you can get the number of frames using
>>> mols.num_frames()
500
and can view the movie using
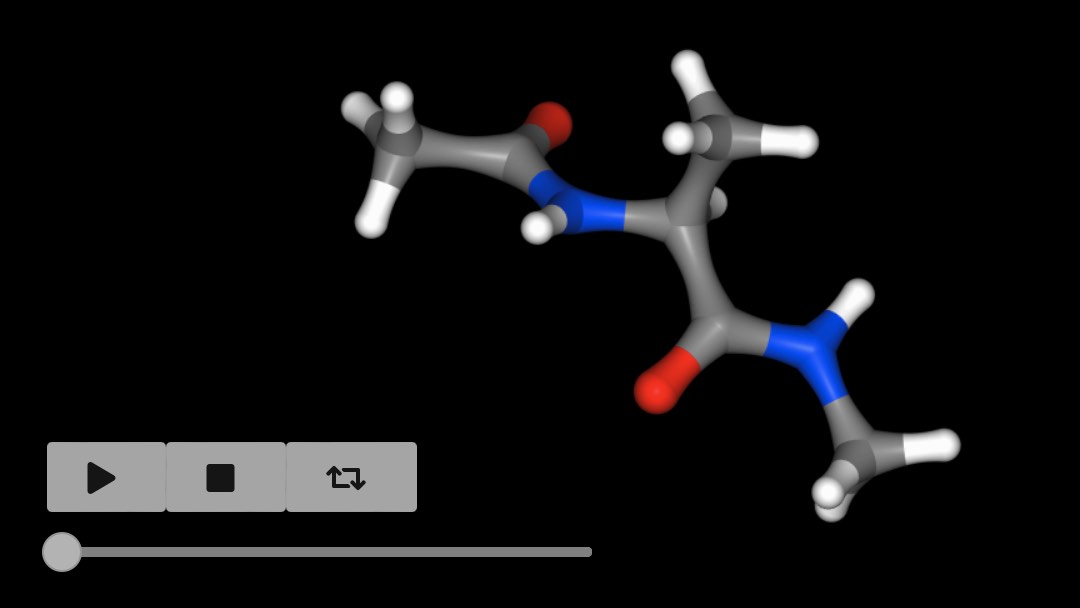
>>> mols.view()

The view() function can be called on any
selection, so you can view the movie of the first molecule using
>>> mols[0].view()
You can extract a subset of trajectory frames by indexing, e.g.
>>> mols[0].trajectory()[0::100].view()
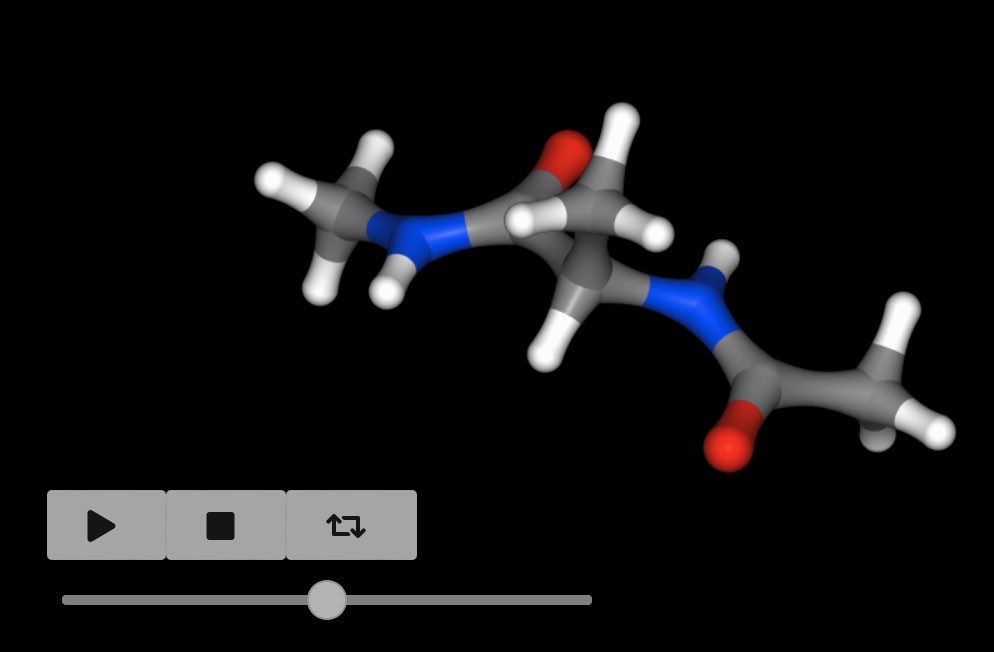
views every 100 frames of the trajectory.
If the molecule was loaded with forcefield parameters, then you can
calculate its energy using the energy()
function.
>>> mols[0].energy()
31.5691 kcal mol-1
You can get all of the components via
>>> mols[0].energy().components()
{'bond': 4.22497 kcal mol-1,
'1-4_LJ': 3.50984 kcal mol-1,
'angle': 7.57006 kcal mol-1,
'dihedral': 9.80034 kcal mol-1,
'1-4_coulomb': 44.8105 kcal mol-1,
'intra_LJ': -1.31125 kcal mol-1,
'improper': 0.485545 kcal mol-1,
'intra_coulomb': -37.5208 kcal mol-1}
You can calculate the energy across a trajectory, with the results returned as a pandas dataframe!
>>> mols[0].trajectory().energy()
frame time 1-4_LJ 1-4_coulomb angle bond dihedral improper intra_LJ intra_coulomb total
0 0 0.200000 3.509838 44.810452 7.570059 4.224970 9.800343 0.485545 -1.311255 -37.520806 31.569147
1 1 0.400000 2.700506 47.698455 12.470519 2.785874 11.776295 1.131481 -1.617496 -40.126219 36.819417
2 2 0.600000 2.801076 43.486411 11.607753 2.023439 11.614774 0.124729 -1.103966 -36.633297 33.920920
3 3 0.800000 3.365638 47.483966 6.524609 0.663454 11.383852 0.339333 -0.983872 -40.197920 28.579061
4 4 1.000000 3.534830 48.596027 6.517530 2.190370 10.214994 0.255331 -1.699613 -40.355054 29.254415
.. ... ... ... ... ... ... ... ... ... ... ...
495 495 99.199997 2.665994 42.866319 11.339087 4.172684 9.875872 0.356887 -1.584092 -36.499764 33.192988
496 496 99.400002 3.062467 44.852774 9.268408 1.878366 10.548897 0.327064 -1.814718 -36.671683 31.451575
497 497 99.599998 3.530233 44.908117 10.487378 4.454670 10.223964 1.006034 -0.692972 -37.118048 36.799376
498 498 99.800003 3.511116 42.976288 9.017446 0.809064 10.841436 0.518190 -1.862433 -35.481467 30.329641
499 499 100.000000 3.768998 41.625135 13.629923 1.089916 11.889372 0.846805 -1.897328 -36.547672 34.405149
You can do more with the molecule, for example viewing it’s 2D structure
>>> mols[0].view2d()
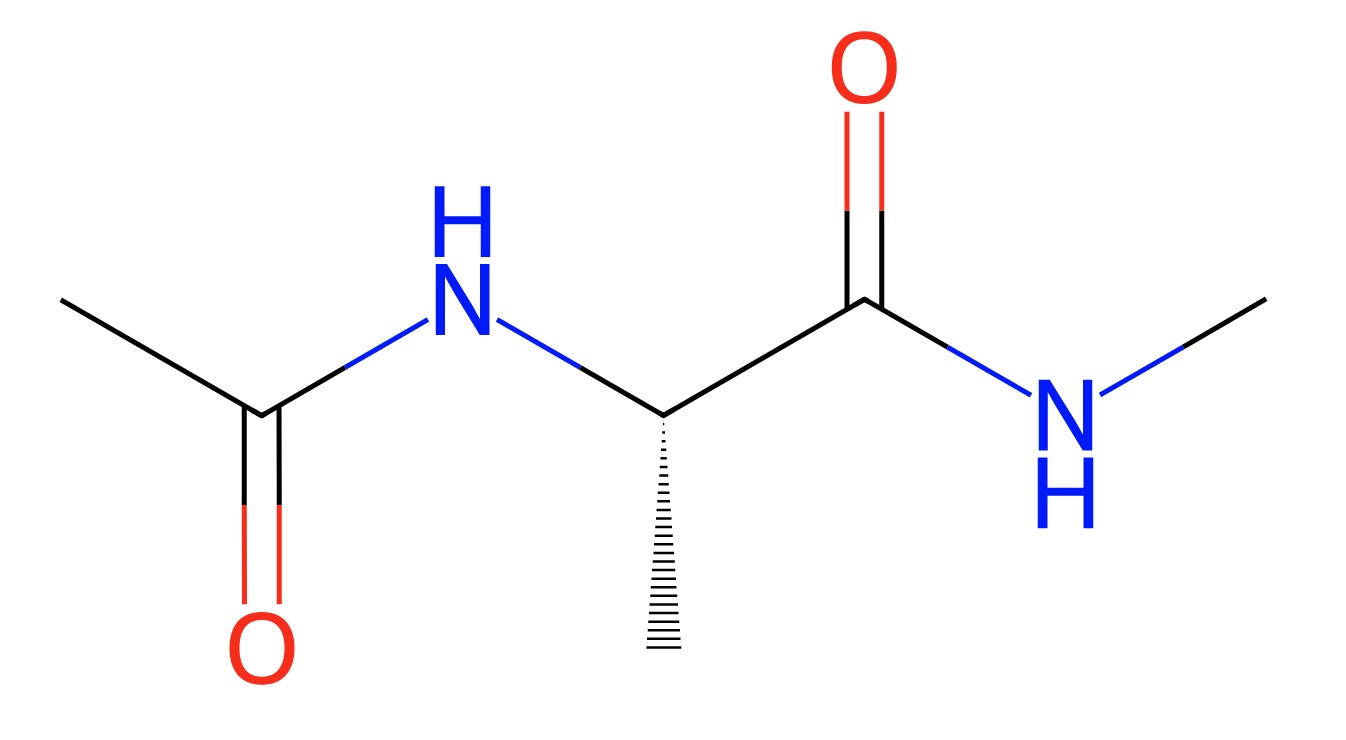
or generating its smiles string.
>>> mols[0].smiles()
'CNC(=O)C(C)NC(C)=O'
You can also convert it to an RDKit molecule!
>>> rdmol = sr.convert.to(mols[0], "rdkit")
And you can even run molecular dynamics using the integration with OpenMM.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "kigaki.gro", "kigaki.top"),
... silent=True)
>>> mols.view()

>>> mols = mols.minimisation().run().commit()
>>> d = mols.dynamics(timestep="4fs")
>>> d.run("20ps", save_frequency="1ps")
>>> mols = d.commit()
>>> mols.trajectory().energy()
frame time 1-4_LJ 1-4_coulomb LJ angle bond coulomb dihedral intra_LJ intra_coulomb total
0 0 1.0 59.828605 1356.012783 9717.588398 34.081250 9.935577e-08 -58901.908280 145.262639 -60.009867 -830.297820 -48479.442291
1 1 2.0 59.859426 1358.426949 9721.478025 33.488730 1.020368e-07 -58977.547695 143.620760 -63.368620 -832.992328 -48557.034752
2 2 3.0 60.891111 1363.801767 9854.057685 32.568284 9.815839e-08 -59169.733258 145.274884 -62.029406 -838.669248 -48613.838181
3 3 4.0 59.832609 1357.912373 9851.460866 32.471396 2.825340e-07 -59192.703528 140.362271 -65.433697 -829.466133 -48645.563842
4 4 5.0 60.074686 1359.927851 9879.077092 34.170306 1.003672e-07 -59240.068442 139.836463 -64.083724 -833.119507 -48664.185275
5 5 6.0 59.375049 1358.601877 9886.713393 34.034265 9.092219e-08 -59270.897562 140.563988 -63.993368 -831.838422 -48687.440781
6 6 7.0 59.614215 1357.914063 9857.271105 34.639254 1.061152e-07 -59250.463890 140.829589 -64.766169 -829.218503 -48694.180337
7 7 8.0 59.926889 1361.405155 9957.054478 34.806703 3.159753e-07 -59380.997407 143.988778 -66.272231 -836.888535 -48726.976172
8 8 9.0 58.966007 1358.311827 9967.327772 34.997531 3.027917e-07 -59405.583408 142.485999 -64.733607 -831.207715 -48739.435594
9 9 10.0 60.324323 1360.640600 9939.400939 36.198475 1.166730e-07 -59388.226935 143.212016 -64.316508 -837.483867 -48750.250956
10 10 11.0 61.320326 1362.023485 9955.071224 34.056823 1.139431e-07 -59424.212266 143.513301 -64.975309 -837.997195 -48771.199611
11 11 12.0 60.415993 1362.776379 9936.669535 31.304641 9.647357e-08 -59410.580161 145.678519 -64.433834 -835.417971 -48773.586899
12 12 13.0 60.970833 1366.508370 9986.881634 33.251273 2.509407e-07 -59466.056471 144.351857 -65.429617 -841.930932 -48781.453053
13 13 14.0 60.245588 1361.266409 9986.122124 34.192312 1.163054e-07 -59480.201397 145.712628 -61.013711 -838.646145 -48792.322190
14 14 15.0 61.878785 1370.377196 9924.323617 31.399821 2.785520e-07 -59418.248563 144.305975 -64.940040 -845.430135 -48796.333344
15 15 16.0 60.430617 1365.739457 9926.082150 33.156124 2.836895e-07 -59423.448702 144.681590 -64.043674 -840.110188 -48797.512625
16 16 17.0 60.294772 1360.174160 9950.578785 32.453372 1.011507e-07 -59450.019119 143.409696 -64.041887 -835.388935 -48802.539157
17 17 18.0 61.156756 1363.542712 9947.586104 34.036896 9.579309e-08 -59452.548714 146.142252 -64.800847 -839.303707 -48804.188547
18 18 19.0 60.048626 1364.033346 9976.885470 33.771207 9.353054e-08 -59483.125256 144.011481 -64.081234 -838.157968 -48806.614328
19 19 20.0 61.609644 1363.227511 9935.113811 32.689491 2.968531e-07 -59452.910379 144.467384 -64.420695 -835.410744 -48815.633975
This integration extends to running GPU-accelerated alchemical molecular dynamics free energy calculations. For example, load up this merged molecule that uses a λ-coordinate to morph between ethane and methanol.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "merged_molecule.s3"))
We’ll now select the reference state (ethane)…
>>> mols = sr.morph.link_to_reference(mols)
To calculate a free energy, we would need to run multiple simulations across λ, calculating the difference in energy between neighbouring λ-windows. How to do this is described in full here. For this quick start guide, we’ll just run at λ=0.5, calculating the difference in energy between λ=0.5 and λ=0.0 and λ=1.0.
First, lets minimise at λ=0.5.
>>> mols = mols.minimisation(lambda_value=0.5).run().commit()
And not lets run a short dynamics simulation at λ=0.5, calculating the energy differences to λ=0.0 and λ=1.0.
>>> d = mols.dynamics(lambda_value=0.5, timestep="1fs", temperature="25oC")
>>> d.run("5ps", energy_frequency="0.5ps", lambda_windows=[0.0, 1.0])
We can now extract the energy differences in an alchemlyb-compatible pandas DataFrame.
>>> print(d.commit().energy_trajectory().to_alchemlyb())
0.0 0.5 1.0
time fep-lambda
0.5 0.5 -46062.581449 -46066.727158 -46064.969441
1.0 0.5 -43335.286994 -43351.114108 -43348.340617
1.5 0.5 -41676.617061 -41685.901393 -41684.741190
2.0 0.5 -40392.020981 -40449.196087 -40448.185263
2.5 0.5 -39720.265484 -39743.531651 -39740.788035
3.0 0.5 -39067.899327 -39094.033563 -39092.723982
3.5 0.5 -38659.468399 -38677.805074 -38676.375989
4.0 0.5 -38354.706210 -38366.022091 -38363.577232
4.5 0.5 -38160.932310 -38175.773525 -38172.820779
5.0 0.5 -37844.757596 -37850.187961 -37848.280865
See the tutorial for a complete script to run and analyse relative free energy calculations.
This is just the beginning of what sire can do! To learn more,
please take a look at the detailed guides
or the tutorial.