Creating merge molecules¶
Merged molecules are used in free energy calculations to represent the perturbation between two molecules; the reference molecule (at λ=0) and the perturbed molecule (at λ=1).
To start, let’s load up two molecules, neopentane and methane, which we will use to create a merged molecule to calculate the relative hydration free energy.
>>> neopentane = sr.load_test_files("neopentane.prm7", "neopentane.rst")[0]
>>> neopentane.view()

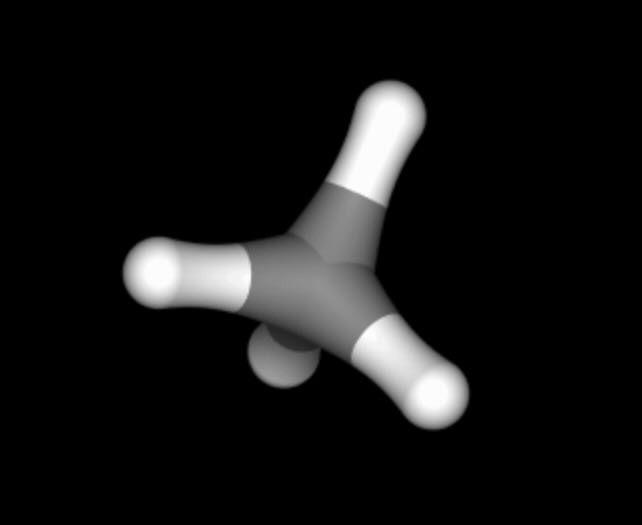
>>> methane = sr.load_test_files("methane.prm7", "methane.rst")[0]
>>> methane.view()
Matching atoms¶
The first step to creating a merged molecule is to decide how atoms should relate between the two end states. This is done by matching atoms from the reference molecule (in this case neopentane) to the perturbed molecule (in this case methane).
For example, let’s say that we want the central carbon of neopentane to perturb into the central carbon of methane. We could specify this by creating a dictionary that says that the name of this atom in neopentane should map to the name of the equivalent atom in methane.
We can get the name of these atoms using the 3D viewer, as shown above.
The central carbon of neopentane is called C1, while the central
carbon of methane is also called C1.
>>> matching = {"C1": "C1"}
Next, we would match each of the other carbon atoms in neopentane to the hydrogen atoms in methane.
>>> matching["C2"] = "H2"
>>> matching["C3"] = "H3"
>>> matching["C4"] = "H4"
>>> matching["C5"] = "H5"
>>> print(matching)
{'C1': 'C1', 'C2': 'H2', 'C3': 'H3', 'C4': 'H4', 'C5': 'H5'}
Merging molecules¶
We use the sire.morph.merge() function to create merged molecules.
This takes in the two molecules you want to merge, and the matching
dictionary you have created.
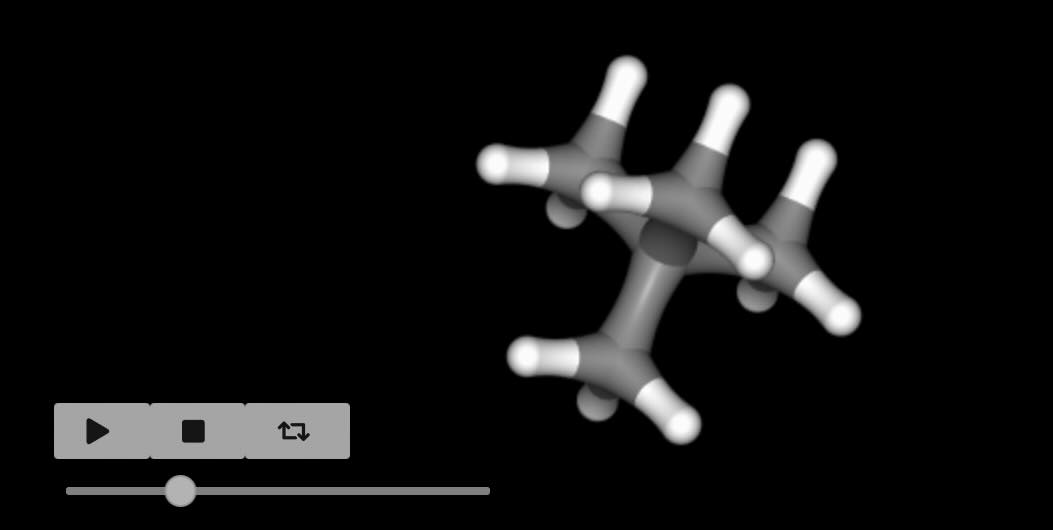
>>> merged = sr.morph.merge(mol0=neopentane, mol1=methane, match=matching)
>>> merged.perturbation().view_reference()
Note
The reference state is numbered 0 (i.e. mol0) while the
perturbed state is numbered 1 (i.e. mol1). This is used to
remind us what λ-value each state corresponds to.
We can see how the underlying OpenMM parameters will be perturbed by
checking the PerturbableOpenMMMolecule
that would be created from the merged molecule.
>>> p = merged.perturbation().to_openmm()
>>> print(p.changed_atoms())
atom charge0 charge1 sigma0 sigma1 epsilon0 epsilon1 alpha0 alpha1 kappa0 kappa1
0 C2:1 -0.085335 0.0271 0.339967 0.264953 0.457730 0.065689 0.0 0.0 0.0 0.0
1 C1:2 -0.060235 -0.1084 0.339967 0.339967 0.457730 0.457730 0.0 0.0 0.0 0.0
2 C3:3 -0.085335 0.0271 0.339967 0.264953 0.457730 0.065689 0.0 0.0 0.0 0.0
3 C4:4 -0.085335 0.0271 0.339967 0.264953 0.457730 0.065689 0.0 0.0 0.0 0.0
4 C5:5 -0.085335 0.0271 0.339967 0.264953 0.457730 0.065689 0.0 0.0 0.0 0.0
5 H6:6 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
6 H7:7 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
7 H8:8 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
8 H9:9 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
9 H10:10 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
10 H11:11 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
11 H12:12 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
12 H13:13 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
13 H14:14 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
14 H15:15 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
15 H16:16 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
16 H17:17 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
We can see here that the five carbons in neopentane have their charge and LJ parameters perturbed from the neopentane values to the methane values.
We can also see that the hydrogens in neopentane are converted to ghost atoms, as they have no match in methane. The conversion to ghost atoms involves setting their charge to zero, and setting the LJ epsilon parameter of the atoms to zero (keeping the sigma parameter the same, as suggested as best practice in the last tutorial).
Note also how the alpha parameters of the hydrogens changes from 0 to 1 as those atoms become ghosts.
We can also look at the changes in internal parameters.
>>> print(p.changed_bonds())
bond length0 length1 k0 k1
0 C2:1-C1:2 0.15375 0.10969 251793.12 276646.08
1 C1:2-C3:3 0.15375 0.10969 251793.12 276646.08
2 C1:2-C4:4 0.15375 0.10969 251793.12 276646.08
3 C1:2-C5:5 0.15375 0.10969 251793.12 276646.08
>>> print(p.changed_angles())
angle size0 size1 k0 k1
0 C4:4-C1:2-C5:5 1.946217 1.877626 526.3472 329.6992
1 C3:3-C1:2-C4:4 1.946217 1.877626 526.3472 329.6992
2 C3:3-C1:2-C5:5 1.946217 1.877626 526.3472 329.6992
3 C2:1-C1:2-C3:3 1.946217 1.877626 526.3472 329.6992
4 C2:1-C1:2-C4:4 1.946217 1.877626 526.3472 329.6992
5 C2:1-C1:2-C5:5 1.946217 1.877626 526.3472 329.6992
>>> print(p.changed_torsions())
Empty DataFrame
Columns: [torsion, k0, k1, periodicity0, periodicity1, phase0, phase1]
Index: []
We can see that the bond lengths and angles are perturbed from their values in neopentane (representing C-C bonds and C-C-C angles) to their values in methane (representing C-H bonds and H-C-H angles). Note that the bonds, angles and torsions for the hydrogens in neopentane are not perturbed. This is because all of these atoms are converted to ghost atoms, and the default is that internals involving ghost atoms keep the parameters from the end state where they are not ghosts (i.e. the reference state values in this case).
Implementation - AtomMapping¶
Under the hood, the above merge was implemented via the
sire.mol.AtomMapping class. This class holds all of the information
about how atoms are mapped between end states, and an object of this
class was created automatically by the merge() function.
We can create the mapping object directly using the
sire.morph.match() function.
>>> m = sr.morph.match(mol0=neopentane, mol1=methane, match=matching)
>>> print(m)
AtomMapping( size=5, unmapped0=12, unmapped1=0
0: MolNum(3) Atom( C1:2 ) <=> MolNum(2) Atom( C1:2 )
1: MolNum(3) Atom( C2:1 ) <=> MolNum(2) Atom( H2:1 )
2: MolNum(3) Atom( C4:4 ) <=> MolNum(2) Atom( H4:4 )
3: MolNum(3) Atom( C3:3 ) <=> MolNum(2) Atom( H3:3 )
4: MolNum(3) Atom( C5:5 ) <=> MolNum(2) Atom( H5:5 )
)
This shows how the five carbon atoms in neopentane are mapped to the
carbon and four hydrogens of methane. It also shows how 12 atoms in the
reference state are unmapped (unmapped0=12) and how no atoms in the
perturbed state are unmapped (unmapped1=0).
This class has some useful functions. One is
align(), which aligns the perturbed state
against the reference state, using an algorithm that minimises the
RMSD of the mapped atoms.
>>> m = m.align()
Another useful function is merge(), which
actually performs the merge, returning the merged molecule, with
perturbable properties linked to the reference state.
>>> merged = m.merge()
>>> print(merged)
Molecule( NEO:7 num_atoms=17 num_residues=1 )
The merge() function is really a wrapper that create
this AtomMapping object, calls align, and then
calls the merge() function.
Automatic matching¶
Creating the matching dictionary by hand can be a bit tedious! Fortunately, there are lots of tools that can help automate this process.
One such tool is the above match() function. If you don’t
pass in a match argument, then an internal maximum common substructure
algorithm will be used to try to infer the matching.
>>> m = sr.morph.match(mol0=neopentane, mol1=methane)
>>> print(m)
AtomMapping( size=1, unmapped0=16, unmapped1=4
0: MolNum(3) Atom( C2:1 ) <=> MolNum(2) Atom( C1:2 )
)
Warning
The maximum common substructure algorithm is not supported on Windows. This will only work on MacOS and Linux.
By default, this algorithm ignores hydrogens. This is why only a single
atom was matched above. You can change this by passing in the
match_light_atoms=True argument.
>>> m = sr.morph.match(mol0=neopentane, mol1=methane, match_light_atoms=True)
>>> print(m)
AtomMapping( size=4, unmapped0=13, unmapped1=1
0: MolNum(3) Atom( C2:1 ) <=> MolNum(2) Atom( C1:2 )
1: MolNum(3) Atom( H6:6 ) <=> MolNum(2) Atom( H2:1 )
2: MolNum(3) Atom( H8:8 ) <=> MolNum(2) Atom( H4:4 )
3: MolNum(3) Atom( H7:7 ) <=> MolNum(2) Atom( H3:3 )
)
Note how this has come up with a different mapping than the one we created manually.
The internal algorithm is quite slow, especially for large molecules. It is also not aware of stereochemistry, and generally not recommended if other tools are available.
Fortunately, because the funtion accepts a python dictionary, it is very easy to use other tools to generate the mapping and pass to this function.
Using Kartograf mappings¶
Kartograf is a package for generating atom mappings which takes into account 3D geometries. This means that mappings can account for stereochemistry. It is also extremely fast and robust, with lots of active development. It is a very good choice for generating atom mappings.
To use Kartograf, you may need to install it. You can do this with conda.
conda install -c conda-forge kartograf
Next, we will import the components of Kartograf that we need.
>>> from kartograf.atom_aligner import align_mol_shape
>>> from kartograf import KartografAtomMapper, SmallMoleculeComponent
Kartograf can work from RDKit molecules, so we’ll now convert our sire molecules to RDKit.
>>> rd_neopentane = sr.convert.to(neopentane, "rdkit")
>>> rd_methane = sr.convert.to(methane, "rdkit")
Next, we will create two Kartograf molecules from these RDKit molecules.
>>> k_neopentane, k_methane = [
... SmallMoleculeComponent.from_rdkit(m) for m in [rd_neopentane, rd_methane]
... ]
Now, we align the molecules based on their shape, aligning methane on top of neopentane.
>>> k_aligned_methane = align_mol_shape(k_methane, ref_mol=k_neopentane)
To generate the mappings, we will create a KartografAtomMapper object which is allowed to match light atoms.
>>> mapper = KartografAtomMapper(atom_map_hydrogens=True)
This can be used to generate the mappings.
>>> mappings = mapper.suggest_mappings(k_neopentane, k_aligned_methane)
We will now get the first mapping…
>>> mapping = next(mappings)
>>> print(mapping)
LigandAtomMapping(componentA=SmallMoleculeComponent(name=NEO),
componentB=SmallMoleculeComponent(name=CH4),
componentA_to_componentB={1: 0, 11: 2, 12: 3, 13: 4, 3: 1},
annotations={})
The mappings are atom indexes. We can collect these and create a dictionary that maps from atom index to atom index using this code.
>>> matching = {}
>>> for atom0, atom1 in mapping.componentA_to_componentB.items():
... matching[sr.atomid(idx=atom0)] = sr.atomid(idx=atom1)
>>> print(matching)
{AtomIdx(1): AtomIdx(0), AtomIdx(11): AtomIdx(2), AtomIdx(12): AtomIdx(3),
AtomIdx(13): AtomIdx(4), AtomIdx(3): AtomIdx(1)}
Finally, we can pass this into the match() function
to create the AtomMapping object.
>>> mapping = sr.morph.match(mol0=neopentane, mol1=methane, match=matching)
>>> print(mapping)
AtomMapping( size=5, unmapped0=12, unmapped1=0
0: MolNum(3) Atom( H12:12 ) <=> MolNum(2) Atom( H3:3 )
1: MolNum(3) Atom( H14:14 ) <=> MolNum(2) Atom( H5:5 )
2: MolNum(3) Atom( H13:13 ) <=> MolNum(2) Atom( H4:4 )
3: MolNum(3) Atom( C1:2 ) <=> MolNum(2) Atom( H2:1 )
4: MolNum(3) Atom( C4:4 ) <=> MolNum(2) Atom( C1:2 )
)
Automatic Kartograf mapping¶
The above code can be quite tedious to write, so we have created the
ability to pass in a KartografAtomMapper object as the match
argument to the match() and merge()
functions, which then does all of the above automatically.
>>> mapper = KartografAtomMapper(atom_map_hydrogens=True)
>>> mapping = sr.morph.match(mol0=neopentane, mol1=methane, match=mapper)
>>> print(mapping)
AtomMapping( size=5, unmapped0=12, unmapped1=0
0: MolNum(3) Atom( H12:12 ) <=> MolNum(2) Atom( H3:3 )
1: MolNum(3) Atom( H14:14 ) <=> MolNum(2) Atom( H5:5 )
2: MolNum(3) Atom( H13:13 ) <=> MolNum(2) Atom( H4:4 )
3: MolNum(3) Atom( C1:2 ) <=> MolNum(2) Atom( H2:1 )
4: MolNum(3) Atom( C4:4 ) <=> MolNum(2) Atom( C1:2 )
)
>>> merged = sr.morph.merge(mol0=neopentane, mol1=methane, match=mapper)
>>> print(merged)
Molecule( NEO:8 num_atoms=17 num_residues=1 )
>>> m = merged.perturbation().to_openmm()
>>> print(m.changed_atoms())
atom charge0 charge1 sigma0 sigma1 epsilon0 epsilon1 alpha0 alpha1 kappa0 kappa1
0 C2:1 -0.085335 0.0000 0.339967 0.339967 0.457730 0.000000 0.0 1.0 1.0 1.0
1 C1:2 -0.060235 0.0271 0.339967 0.264953 0.457730 0.065689 0.0 0.0 0.0 0.0
2 C3:3 -0.085335 0.0000 0.339967 0.339967 0.457730 0.000000 0.0 1.0 1.0 1.0
3 C4:4 -0.085335 -0.1084 0.339967 0.339967 0.457730 0.457730 0.0 0.0 0.0 0.0
4 C5:5 -0.085335 0.0000 0.339967 0.339967 0.457730 0.000000 0.0 1.0 1.0 1.0
5 H6:6 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
6 H7:7 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
7 H8:8 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
8 H9:9 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
9 H10:10 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
10 H11:11 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
11 H12:12 0.033465 0.0271 0.264953 0.264953 0.065689 0.065689 0.0 0.0 0.0 0.0
12 H13:13 0.033465 0.0271 0.264953 0.264953 0.065689 0.065689 0.0 0.0 0.0 0.0
13 H14:14 0.033465 0.0271 0.264953 0.264953 0.065689 0.065689 0.0 0.0 0.0 0.0
14 H15:15 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
15 H16:16 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
16 H17:17 0.033465 0.0000 0.264953 0.264953 0.065689 0.000000 0.0 1.0 1.0 1.0
This is a much easier way to create merged molecules!
Extracting the end states¶
As with all merged molecules, we can extract the end states via the
sire.morph.extract_reference() and sire.morph.extract_perturbed()
functions.
>>> neopentane = sr.morph.extract_reference(merged)
>>> print(neopentane.atoms())
Selector<SireMol::Atom>( size=17
0: Atom( C2:1 [ 2.24, 1.01, 0.00] )
1: Atom( C1:2 [ 1.26, 0.09, -0.75] )
2: Atom( C3:3 [ 0.99, 0.66, -2.15] )
3: Atom( C4:4 [ -0.06, -0.00, 0.04] )
4: Atom( C5:5 [ 1.88, -1.32, -0.88] )
...
12: Atom( H13:13 [ -0.78, -0.67, -0.49] )
13: Atom( H14:14 [ 0.11, -0.42, 1.05] )
14: Atom( H15:15 [ 2.83, -1.27, -1.44] )
15: Atom( H16:16 [ 2.08, -1.75, 0.13] )
16: Atom( H17:17 [ 1.19, -2.00, -1.42] )
)
>>> methane = sr.morph.extract_perturbed(merged)
>>> print(methane.atoms())
Selector<SireMol::Atom>( size=5
0: Atom( H2:2 [ -0.45, 1.01, 0.10] )
1: Atom( C1:4 [ -0.00, 0.00, 0.00] )
2: Atom( H3:12 [ -0.71, -0.67, -0.53] )
3: Atom( H4:13 [ 0.95, 0.07, -0.57] )
4: Atom( H5:14 [ 0.21, -0.41, 1.01] )
)
The names of the atoms and residues are preserved in the merged molecule, meaning that they are correctly output for each end state. In the merged molecule, the atom and residue names default to the reference state names. The perturbed state names are held in the “alternate” names.
>>> print(merged.residues()[0].name(), merged.residues()[0].alternate_name())
ResName('NEO') ResName('CH4')
>>> print(merged[1].name(), merged[1].alternate_name())
AtomName('C1') AtomName('H2')
Atoms that are unmapped in an end state are called Xxx, e.g.
>>> print(merged[0].name(), merged[0].alternate_name())
AtomName('C2') AtomName('Xxx')
You can switch between the standard and alternate names of a molecule
by calling the switch_to_alternate_names()
function.
>>> print(merged.atoms())
Selector<SireMol::Atom>( size=17
0: Atom( C2:1 [ 2.24, 1.01, 0.00] )
1: Atom( C1:2 [ 1.26, 0.09, -0.75] )
2: Atom( C3:3 [ 0.99, 0.66, -2.15] )
3: Atom( C4:4 [ -0.06, -0.00, 0.04] )
4: Atom( C5:5 [ 1.88, -1.32, -0.88] )
...
12: Atom( H13:13 [ -0.78, -0.67, -0.49] )
13: Atom( H14:14 [ 0.11, -0.42, 1.05] )
14: Atom( H15:15 [ 2.83, -1.27, -1.44] )
15: Atom( H16:16 [ 2.08, -1.75, 0.13] )
16: Atom( H17:17 [ 1.19, -2.00, -1.42] )
)
>>> merged = merged.edit().switch_to_alternate_names().commit()
>>> print(merged.atoms())
Selector<SireMol::Atom>( size=17
0: Atom( Xxx:1 [ 2.24, 1.01, 0.00] )
1: Atom( H2:2 [ 1.26, 0.09, -0.75] )
2: Atom( Xxx:3 [ 0.99, 0.66, -2.15] )
3: Atom( C1:4 [ -0.06, -0.00, 0.04] )
4: Atom( Xxx:5 [ 1.88, -1.32, -0.88] )
...
12: Atom( H4:13 [ -0.78, -0.67, -0.49] )
13: Atom( H5:14 [ 0.11, -0.42, 1.05] )
14: Atom( Xxx:15 [ 2.83, -1.27, -1.44] )
15: Atom( Xxx:16 [ 2.08, -1.75, 0.13] )
16: Atom( Xxx:17 [ 1.19, -2.00, -1.42] )
)
>>> merged = merged.edit().switch_to_alternate_names().commit()
>>> print(merged.atoms())
Selector<SireMol::Atom>( size=17
0: Atom( C2:1 [ 2.24, 1.01, 0.00] )
1: Atom( C1:2 [ 1.26, 0.09, -0.75] )
2: Atom( C3:3 [ 0.99, 0.66, -2.15] )
3: Atom( C4:4 [ -0.06, -0.00, 0.04] )
4: Atom( C5:5 [ 1.88, -1.32, -0.88] )
...
12: Atom( H13:13 [ -0.78, -0.67, -0.49] )
13: Atom( H14:14 [ 0.11, -0.42, 1.05] )
14: Atom( H15:15 [ 2.83, -1.27, -1.44] )
15: Atom( H16:16 [ 2.08, -1.75, 0.13] )
16: Atom( H17:17 [ 1.19, -2.00, -1.42] )
)