Viewing Molecules in 2D¶
The ability to convert sire objects to native objects of other
molecular packages makes it easy to create convenience functions that
leverage the capabilities of those packages under the hood.
For example, RDKit can be used to create 2D views of molecules.
This functionality is used by the view2d() function to
create a two-dimensional view of any Molecule,
molecule sub-view or collections of molecules.
In a jupyter notebook you can run;
>>> import sire as sr
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.crd", "ala.top"))
>>> mols[0].view2d()
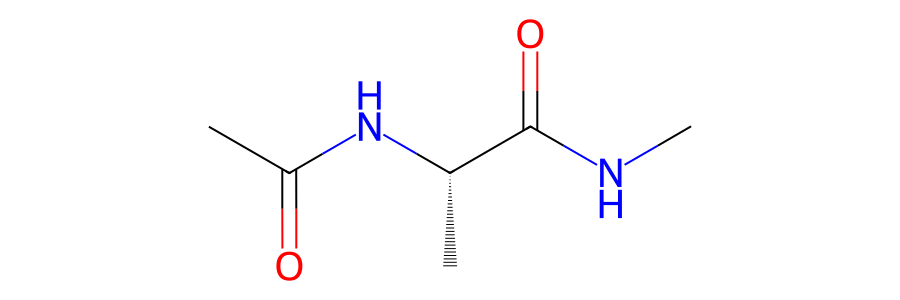
This will render the image to the screen. You can save this to a PNG,
PDF or SVG file using the filename argument, e.g.
>>> mols[0].view2d(filename="aladip.png")
'/path/to/aladip.png'
This returns the full path to the image file created.
The choice of format is based on the extension used for the filename,
e.g. .png for PNG, .pdf for PDF and .svg for SVG.
Note
You may need to have the cairosvg package installed to save
to PNG or PDF format. You will see a warning printed to the screen
if this package is needed, and the image will instead be saved
in SVG format. You can install cairosvg using
conda install cairosvg.
You can control the height and width of the image using the height
and width arguments (measured in pixels), e.g.
>>> mols[0].view2d(filename="small.pdf", height=200, width=200)
'/path/to/small.pdf'
By default, this will not show unambiguous hydrogens (e.g. those that
are not implied by the 2D structure). You can view all hydrogens
using the include_hydrogens option.
>>> mols[0].view2d(include_hydrogens=True)
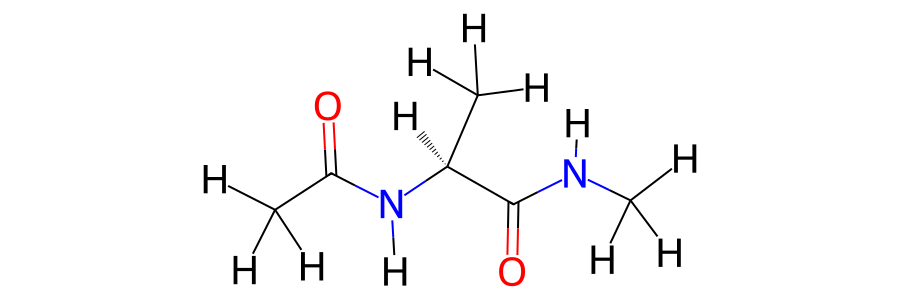
Notice how the stereochemistry, formal charges and bonding are determined automatically for the molecule. This is based on a simple algorithm that add and removes bonds and charges until the valence shell of each atom is full. The stereochemistry is derived based on the 3D structure of the molecule, using functionality built into RDKit.
You can view whole molecules or parts of molecules in 2D. For example, this will view the second residue of the first molecule.
>>> mols[0]["residx 1"].view2d(include_hydrogens=True)
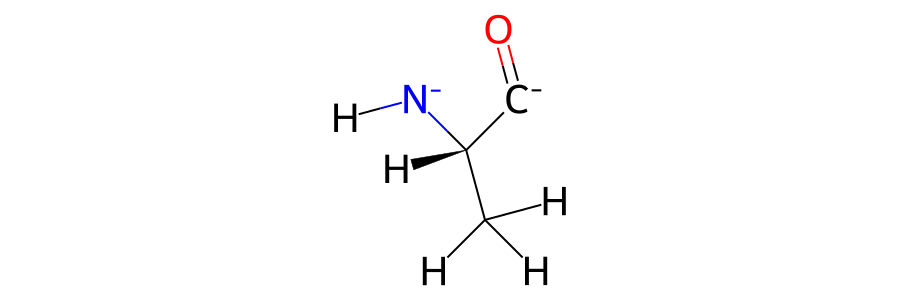
Note
The formal charge and bonding around the atoms that are missing bonds is likely to be incorrect. Here you can see that the nitrogen and carbon have gained negative charges as they have lost their bonds to the neighbouring residues.
You can also view lots of molecules at once in 2D. In this case, molecules are grouped, with only the different molecules contained in the collection shown, with their counts and smiles strings.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "kigaki.gro", "kigaki.top"),
... show_warnings=False)
>>> mols.view2d()
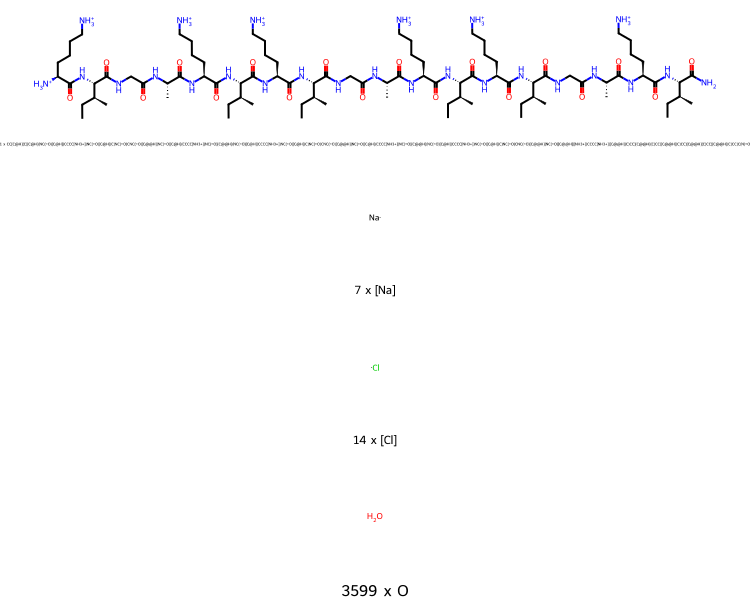
From this you can see that there was one short peptide, with 7 sodium ions, 14 chloride ions and 3599 water molecules.
Note
We do not recommend that you try to create 2D views of large molecules
(e.g. proteins). We plan to add functionality to detect proteins and
instead print out their sequences when viewed via the view2d()
function. Do get in touch if you want to discuss alternative ideas,
or if you want to help contribute to the code.