Merged Molecules¶
The concept of a “merged molecule” is central to the way that free energy calculations are implemented in sire. A merged molecule is one that represents both a “reference” state and a “perturbed” state. These are the two states that the free energy simulation will morph between, and for which the free energy difference will be calculated.
For example, here we have pre-prepared a merged molecule that represents the perturbation from ethane to methanol.
>>> import sire as sr
>>> mols = sr.load(sr.expand(sr.tutorial_url, "merged_molecule.s3"))
>>> print(mols)
System( name=BioSimSpace_System num_molecules=4054 num_residues=4054 num_atoms=12167 )
Note
The .s3 file format is an internal binary format used by sire to
store any object. You can create .s3 files using the
sire.stream.save() function, and load them using the
sire.stream.load() function. These files have the .s3
suffix when created by sire, and the .bss suffix when created
by BioSimSpace. The .s3 format is designed to
be portable and backwards compatible, but standard file formats,
e.g. .pdb, .mol2, .sdf, etc. should be preferred for
long-term storage of data.
Note
This merged molecule was created using BioSimSpace,
and then saved using sire.stream.save().
You should use BioSimSpace if you want to create merged molecules
yourself.
This system contains a single merged molecule in a box of water. Merged
molecules are idenfitied by the molecule property is_perturbable, which
will be True. We can extract the merged molecule from this system using
>>> mol = mols["molecule property is_perturbable"]
>>> print(mol)
Molecule( Merged_Molecule:6 num_atoms=8 num_residues=1 )
A merged molecule is exactly the same as all other molecules in sire. The
difference is that it contains two set of the molecular properties;
one that represents the reference state, and one that represents the
perturbed state. These are identified by the 0 and 1 suffixes.
For example, the reference state coordinates are in the coordinates0
property;
>>> print(mol.property("coordinates0"))
AtomCoords( size=8
0: (25.71278789630378, 24.93752746353058, 25.253932968775896)
1: (24.28721210369622, 25.062578848992636, 24.746067031224108)
2: (25.911542134040474, 23.88985958968847, 25.56394874111798)
3: (26.425045597240814, 25.22062162179178, 24.45094936681738)
4: (25.86160072175123, 25.60943815695771, 26.125882372721225)
5: (24.13839927824877, 24.3906681555655, 23.87411762727878)
6: (24.088796970412663, 26.110140410311534, 24.43511653656108)
7: (23.574954402759186, 24.779484690731437, 25.549050633182624)
)
while the perturbed state coordinates are in the coordinates1 property;
>>> print(mol.property("coordinates1"))
AtomCoords( size=8
0: (25.65553270521631, 24.945670198487242, 25.22503902796385)
1: (24.34753270521631, 25.064670198487246, 24.744039027963847)
2: (25.911542134040474, 23.88985958968847, 25.56394874111798)
3: (26.247532705216308, 25.207670198487243, 24.474039027963848)
4: (25.86160072175123, 25.60943815695771, 26.125882372721225)
5: (24.194532705216307, 24.386670198487245, 23.877039027963846)
6: (24.14453270521631, 26.114670198487243, 24.441039027963846)
7: (23.63753270521631, 24.781670198487245, 25.548039027963846)
)
Similarly the reference state atomic charges are in the charge0 property;
>>> print(mol.property("charge0"))
SireMol::AtomCharges( size=8
0: -0.09435 |e|
1: -0.09435 |e|
2: 0.03145 |e|
3: 0.03145 |e|
4: 0.03145 |e|
5: 0.03145 |e|
6: 0.03145 |e|
7: 0.03145 |e|
)
while the perturbed state atomic charges are in the charge1 property;
>>> print(mol.property("charge1"))
SireMol::AtomCharges( size=8
0: -0.5988 |e|
1: 0.1167 |e|
2: 0 |e|
3: 0.396 |e|
4: 0 |e|
5: 0.0287 |e|
6: 0.0287 |e|
7: 0.0287 |e|
)
The atomic elements are in the element0 and element1 properties;
>>> print(mol.property("element0"))
SireMol::AtomElements( size=8
0: Carbon (C, 6)
1: Carbon (C, 6)
2: Hydrogen (H, 1)
3: Hydrogen (H, 1)
4: Hydrogen (H, 1)
5: Hydrogen (H, 1)
6: Hydrogen (H, 1)
7: Hydrogen (H, 1)
)
>>> print(mol.property("element1"))
SireMol::AtomElements( size=8
0: Oxygen (O, 8)
1: Carbon (C, 6)
2: dummy (Xx, 0)
3: Hydrogen (H, 1)
4: dummy (Xx, 0)
5: Hydrogen (H, 1)
6: Hydrogen (H, 1)
7: Hydrogen (H, 1)
)
Here we can see that the atoms at indexes 2 and 4 go from being hydrogens in the reference state (with charges of 0.03145 |e|) to being ghost (or dummy) atoms in the perturbed state, with charges of zero.
Viewing merged molecules¶
The standard view() function uses the standard
coordinates, element and other properties to view molecules. These
properties don’t exist in our merged molecule, as instead we have
coordinates0, coordinates1, element0, element1, etc.
To view the molecule, we need to choose which of the reference or perturbed
states we want to view. We do this by linking the standard properties to
either the reference or perturbed versions, e.g. linking coordinates
to coordinates0 if we want to view the reference state.
We could do this manually, but it would be a bit tedious. To save typing,
sire provides a sire.morph.Perturbation class that makes it easier
to work with merged molecules. You can access this via the
perturbation() method.
>>> pert = mol.perturbation()
>>> print(pert)
Perturbation( Molecule( Merged_Molecule:6 num_atoms=8 num_residues=1 ) )
Note
Calling the perturbation() method on a molecule
that is not a merged molecule will raise an exception.
The Perturbation class provides the
link_to_reference() and
link_to_perturbed() methods. These can
be used to link all of the standard properties to either the reference
or perturbed values.
>>> mol = pert.link_to_reference()
>>> mol.view()
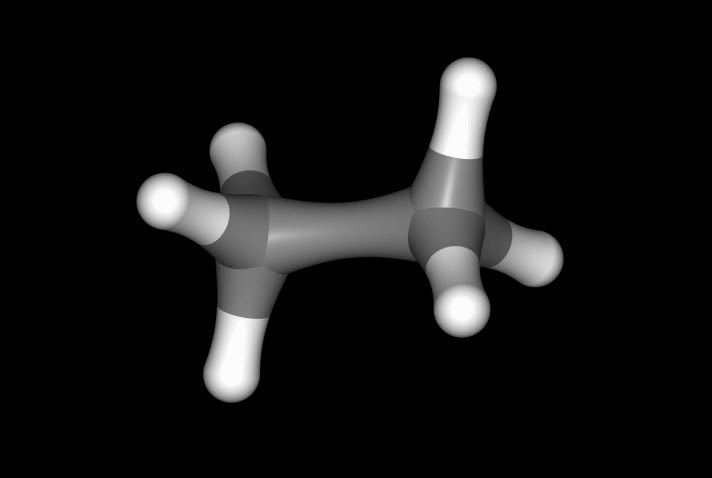
has viewed the reference state (ethane), while
>>> mol = pert.link_to_perturbed()
>>> mol["not element Xx"].view()
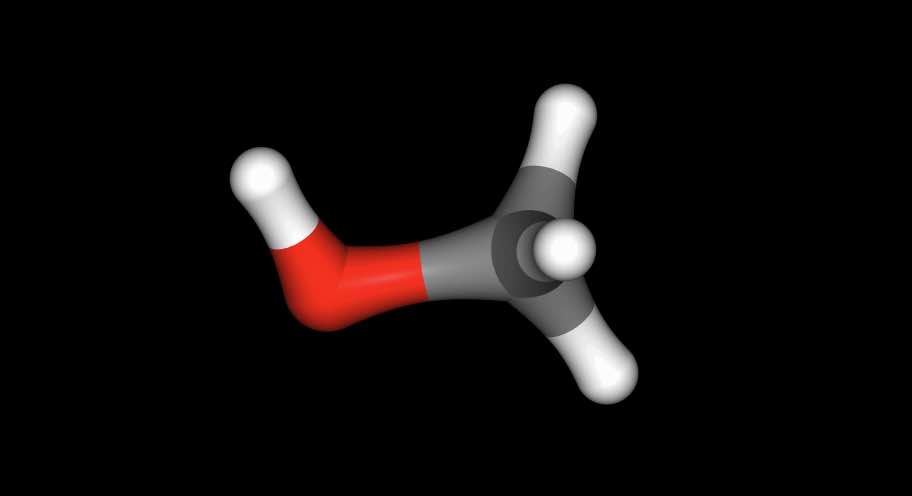
has viewed the perturbed state (methanol).
Note
The perturbed state includes two ghost (dummy) atoms, which should
normally be invisible. However, the view function will show all
atoms, including ghosts. To hide the ghost atoms, we have chosen
to view all non-ghost atoms, i.e. all atoms that are not element
Xx.
Viewing merged molecules in their environment¶
So far we have just viewed the merged molecule in isolation. However, the molecule exists in a system, in this case, a box of water. We can view the merged molecule in its environment by updating the system with the result of linking the molecule to either the reference or perturbed states, e.g.
>>> mols = mols.update(pert.link_to_reference())
has updated the system with a copy of the merged molecule where all of its standard properties are linked to the reference state. While
>>> mols = mols.update(pert.link_to_perturbed())
updates the system with a copy of the merged molecule where all of its standard properties are linked to the perturbed state.
In general, a system could contain many merged molecules. To link all of them to the reference state you could use
>>> for mol in mols.molecules("molecule property is_perturbable"):
... mols.update(mol.perturbation().link_to_reference())
or to link all of them to the perturbed state you could use
>>> for mol in mols.molecules("molecule property is_perturbable"):
... mols.update(mol.perturbation().link_to_perturbed())
The sire.morph.link_to_reference() and
sire.morph.link_to_perturbed() convenience function can do this
for you, e.g.
>>> mols = sr.morph.link_to_reference(mols)
or
>>> mols = sr.morph.link_to_perturbed(mols)
Now you could view and manipulate them as normal, e.g. using
mols.view() etc.