Alchemical Restraints¶
You can perturb restraints as part of an alchemical free energy simulation. This is useful for computing free energy differences between two systems where you want to turn on or off restraints that are used to keep part of the molecules in place while you are performing the mutation.
You can perturb restraints by adding them to the
LambdaSchedule used to control the perturbation.
By default, restraints are called restraint, and so are perturbed
using the restraint lever.
>>> import sire as sr
>>> l = sr.cas.LambdaSchedule()
>>> l.add_stage("restraints_on", l.initial())
>>> l.add_stage("morph", (1-l.lam()) * l.initial() + l.lam() * l.final())
>>> l.add_stage("restraints_off", l.final())
>>> print(l)
LambdaSchedule(
restraints_on: initial
morph: initial * (-λ + 1) + λ * final
restraints_off: final
)
This has created a schedule with three stages, restraints_on, morph
and restraints_off. The morph stage is the one that will be used
to peturb the molecule from the initial state to the perturbed state.
The restraints_on and restraints_off stages will be used to
switch on and off the restraints. To actually switch them on and off,
we need to set the equation that will be used to scale the restraints.
>>> l.set_equation("restraints_on", "restraint", l.lam() * l.initial())
>>> l.set_equation("restraints_off", "restraint", (1-l.lam()) * l.initial())
This will scale the restraints by λ in the restraints_on stage. This
means that the restraints start at λ=0 scaled to 0, and are then fully
switched on by λ=1.
The restraints are scaled off by (1-λ) in the restraints_off stage.
This means that the restraints are fully switched on at λ=0, but then
scaled down so that they are scaled to 0 at λ=1.
The value of initial and final for a restraint is the same, being
100% of the restraint. Thus (1-l.lam()) * l.initial() + l.lam() * l.final()
will always evaluate to 100% of the restraint for all values of λ during
the morph stage. You can make sure that the restraint is kept at
100% during this stage by setting this value explicitly;
>>> l.set_equation(stage="morph", lever="restraint", l.initial())
>>> print(l)
LambdaSchedule(
restraints_on: initial
restraint: λ * initial
morph: initial * (-λ + 1) + λ * final
restraint: initial
restraints_off: final
restraint: final * (-λ + 1)
)
You can view the effect of this schedule by plotting the perturbation
assuming an initial value of 1.0 and a final value of 2.0 using
>>> l.get_lever_values(initial=1.0, final=2.0).plot()
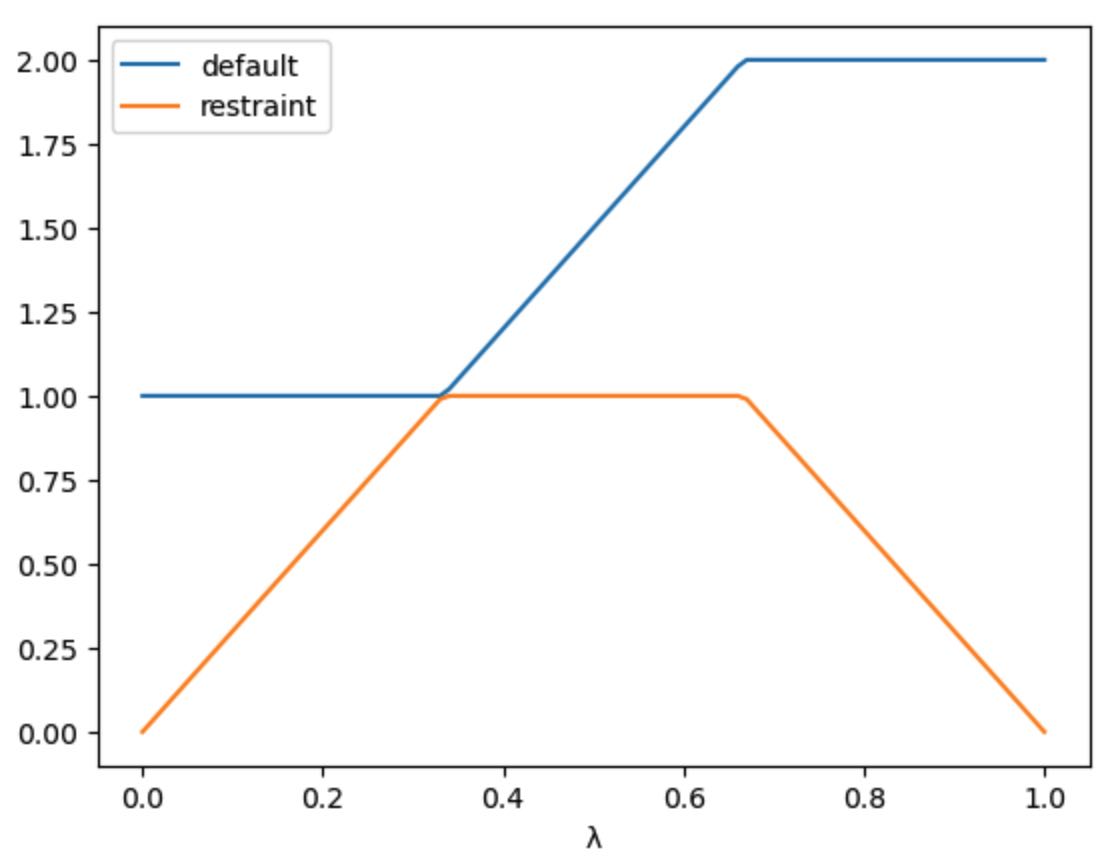
Here we can see the three stages of the schedule. The restraints_on
stage keeps the default value of the restraint at the initial value
(1.0) while it scales the restraint from 0 to 1.0. The morph
stage keeps the restraint at 1.0, while scaling the parameter from
1.0 to 2.0. Finally, the restraints_off stage scales the
restraint from 1.0 to 0 while keeping the parameter at 2.0.
We can now use this schedule to perturb the restraints during an alchemical simulation e.g.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "merged_molecule.s3"))
>>> restraints = sr.restraints.positional(mols, "molidx 0")
>>> mols = mols.minimisation(restraints=restraints,
... lambda_value=0.0).run().commit()
>>> d = mols.dynamics(timestep="4fs", temperature="25oC",
... restraints=restraints, schedule=l,
... lambda_value=0.0)
>>> d.run("10ps")
>>> mols = d.commit()
Using named restraints¶
By default, all restraints in a system are called restraint, and so are
perturbed using the restraint lever. However, you can also give restraints
their own name, and then perturb them using their name. For example, here
we create two restraints, named positional and distance.
>>> pos_rest = sr.restraints.positional(mols, "molidx 0", name="positional")
>>> dst_rest = sr.restraints.distance(mols, atoms0=0, atoms1=1, name="distance")
>>> print(pos_rest, dst_rest)
PositionalRestraints( name=positional, size=8
0: PositionalRestraint( 0 => ( 25.7128, 24.9375, 25.2539 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
1: PositionalRestraint( 1 => ( 24.2872, 25.0626, 24.7461 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
2: PositionalRestraint( 2 => ( 25.9115, 23.8899, 25.5639 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
3: PositionalRestraint( 3 => ( 26.425, 25.2206, 24.4509 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
4: PositionalRestraint( 4 => ( 25.8616, 25.6094, 26.1259 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
5: PositionalRestraint( 5 => ( 24.1384, 24.3907, 23.8741 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
6: PositionalRestraint( 6 => ( 24.0888, 26.1101, 24.4351 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
7: PositionalRestraint( 7 => ( 23.575, 24.7795, 25.5491 ), k=150 kcal mol-1 Å-2 : r0=0 Å )
) BondRestraints( name=distance, size=1
0: BondRestraint( 0 <=> 1, k=150 kcal mol-1 Å-2 : r0=1.5185 Å )
)
We can now create a schedule that perturbs these restraints separately using
their names. We will first scale up the distance restraint in a
distance_restraints stage…
>>> l = sr.cas.LambdaSchedule()
>>> l.add_stage("distance_restraints", 0)
>>> l.set_equation(stage="distance_restraints", lever="distance",
... equation=l.lam() * l.initial())
and will then scale up the positional restraint in a
positional_restraints stage, while keeping the distance restraint
fully on.
>>> l.add_stage("positional_restraints", 1)
>>> l.set_equation(stage="positional_restraints", lever="positional",
... equation=l.lam() * l.initial())
>>> print(l)
LambdaSchedule(
distance_restraints: 0
distance: λ * initial
positional_restraints: 1
positional: λ * initial
)
>>> l.get_lever_values(initial=1.0, final=1.0,
... levers=["positional", "distance"]).plot()
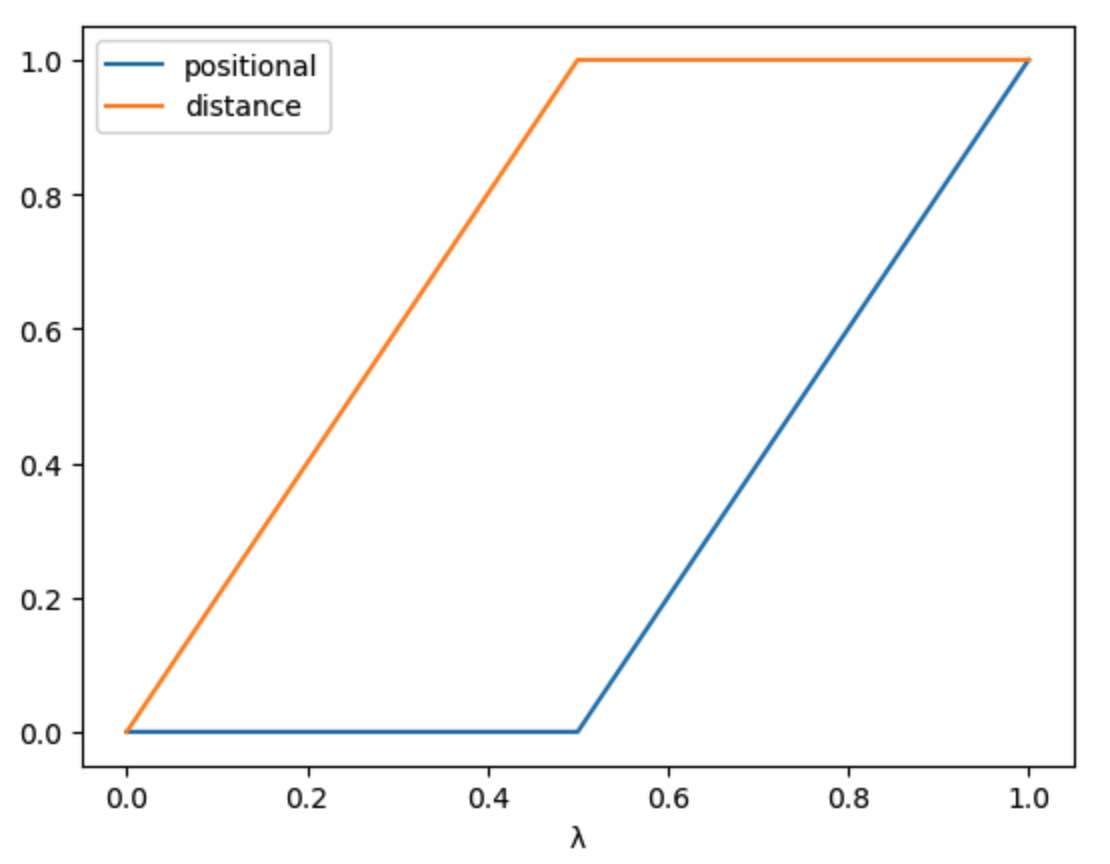
Here we can see that, in the first distance_restraints stage
(from λ=0 to λ=0.5), the distance restraint is scaled from 0 to 1
while the positional restraint is kept at 0. In the second
positional_restraints stage (from λ=0.5 to λ=1), the positional
restraint is scaled from 0 to 1 while the distance restraint is
kept at 1.
We can now use this schedule in a simulation, e.g.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "merged_molecule.s3"))
>>> mols = mols.minimisation(restraints=[dst_rest, pos_rest],
... lambda_value=0.0).run().commit()
>>> d = mols.dynamics(timestep="4fs", temperature="25oC",
... restraints=[dst_rest, pos_rest], schedule=l,
... lambda_value=0.0)
>>> d.run("10ps")
>>> mols = d.commit()