Part 2 - Indexing and Searching¶
The core of sire are the various MoleculeView-derived
classes, such as Atom, Residue,
Chain, Segment and
Molecule, amongst others.
These can all be considered as containers for molecular information.
Atom is a container for atomic information,
Molecule is a container for molecular information etc.
We access this information by indexing or searching these containers, which we will learn how to do in this part of the tutorial.
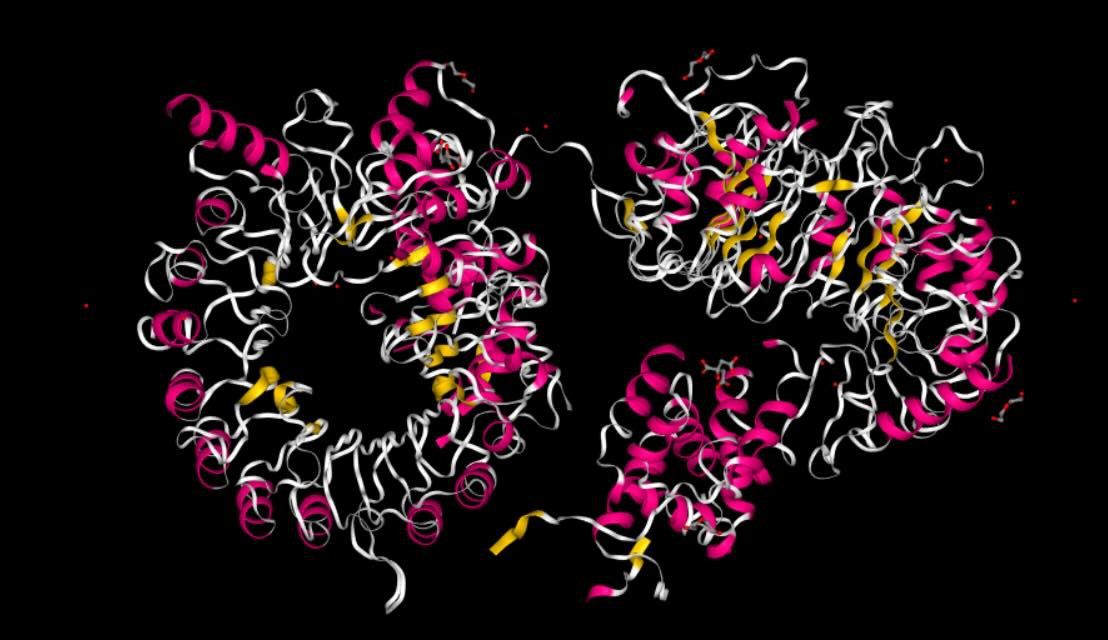
First, let’s load up an example protein,
7SA1 from the PDB
>>> import sire as sr
>>> mols = sr.load("7SA1")
Downloading from 'https://files.rcsb.org/download/7SA1.cif.gz'...
Unzipping './7SA1.cif.gz'...
>>> print(mols)
System( name=7SA1 num_molecules=26 num_residues=1518 num_atoms=11728 )
>>> mols.view()
Note
sire automatically downloads and unpacks structures from the PDB. Just
put in the PDB code as the argument to sire.load().
Note
A new-format PDBx/mmCIF file will be downloaded if you have `gemmi <>`__ installed. Otherwise, a legacy-format PDB file will be downloaded.
Molecules are constructed as atoms, which be can be (optionally) arranged into residues, chains and segments. We can get the number of each using
>>> print(f"The number of atoms is {mols.num_atoms()}")
The number of atoms is 11728
>>> print(f"The number of residues is {mols.num_residues()}")
The number of residues is 1518
>>> print(f"The number of chains is {mols.num_chains()}")
The number of chains is 4
>>> print(f"The number of segments is {mols.num_segments()}")
The number of segments is 0
>>> print(f"The number of molecules is {mols.num_molecules()}")
26