Creating Molecules from Smiles Strings¶
Smiles strings provide a convenient way to represent a molecule as text.
You can create a molecule from a smiles string using the
sire.smiles() function.
>>> import sire as sr
>>> mol = sr.smiles("C1:C:C:C:C:C1")
>>> print(mol.atoms())
Selector<SireMol::Atom>( size=12
0: Atom( C1:1 [ -0.92, -1.05, 0.02] )
1: Atom( C2:2 [ -1.37, 0.27, -0.04] )
2: Atom( C3:3 [ -0.45, 1.32, -0.06] )
3: Atom( C4:4 [ 0.92, 1.05, -0.02] )
4: Atom( C5:5 [ 1.37, -0.27, 0.04] )
...
7: Atom( H8:8 [ -2.43, 0.48, -0.07] )
8: Atom( H9:9 [ -0.80, 2.35, -0.11] )
9: Atom( H10:10 [ 1.63, 1.87, -0.04] )
10: Atom( H11:11 [ 2.43, -0.48, 0.07] )
11: Atom( H12:12 [ 0.80, -2.35, 0.11] )
)
>>> mol.view()
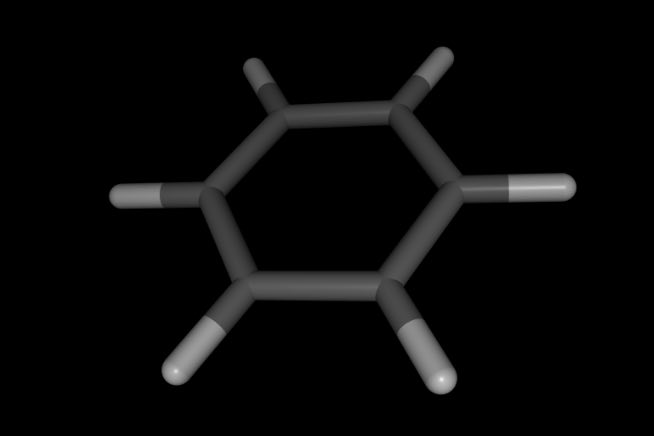
Note how hydrogen atoms and coordinates of all atoms have
been generated automatically. You can control this using
the add_hydrogens and generate_coordinates options, e.g.
>>> mol = sr.smiles("C1:C:C:C:C:C1", generate_coordinates=False)
>>> print(mol.atoms())
Selector<SireMol::Atom>( size=12
0: Atom( C1:1 )
1: Atom( C2:2 )
2: Atom( C3:3 )
3: Atom( C4:4 )
4: Atom( C5:5 )
...
7: Atom( H8:8 )
8: Atom( H9:9 )
9: Atom( H10:10 )
10: Atom( H11:11 )
11: Atom( H12:12 )
)
>>> mol = sr.smiles("C1:C:C:C:C:C1", add_hydrogens=False)
>>> print(mol.atoms())
Selector<SireMol::Atom>( size=6
0: Atom( C1:1 )
1: Atom( C2:2 )
2: Atom( C3:3 )
3: Atom( C4:4 )
4: Atom( C5:5 )
5: Atom( C6:6 )
)
Note
Note that coordinates cannot be generated if
add_hydrogens is False
The above code works by using the
Chem.MolFromSmiles
function from rdkit. This is used to
create an rdkit Molecule
which is converted to a sire Molecule using
the functions in the convert module.
Note
Note that the default is that all sanitization steps provided by rdkit
will be performed when generating the molecule. If any fail, then
an exception will be raised and the molecule won’t be generated.
You can change this behaviour by passing must_sanitize as False.
This will run as many of the sanitization steps as possible, ignoring
errors caused by individual steps.
Generating smiles strings from molecules¶
We can also convert in the other direction, e.g. from a Molecule
to an rdkit Molecule.
This can be used with rdkit’s MolToSmiles
function to generate a smiles string. This is performed automatically
using a molecule’s .smiles() function, e.g.
>>> mol = sr.smiles("C1:C:C:C:C:C1")
>>> print(mol.smiles())
c1ccccc1
Note how hydrogens have been left out from the smiles string. They are only included if they are needed to resolve any ambiguity in the structure or chirality. For example;
>>> mol = sr.smiles("C[C@H](N)C(=O)O")
>>> print(mol.smiles())
C[C@H](N)C(=O)O
You can ask for all of the hydrogens to be included explicitly by
passing include_hydrogens as True.
>>> print(mol.smiles(include_hydrogens=True))
[H]OC(=O)[C@@]([H])(N([H])[H])C([H])([H])[H]
The smiles function can be called on any molecule, even if it hasn’t been
created from a smiles string, e.g.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.crd", "ala.top"))
>>> print(mols[0].smiles())
CNC(=O)C(C)NC(C)=O
You can also call it on a subset of the molecule, e.g.
>>> print(mols[0]["residx 0"].smiles())
C[C-]=O
Note
Note that smiles strings of subsets will have missing bonds, e.g. here we can see that the central carbon has a negative charge because it is missing the bond to the carbon in the next residue.
You can also create smiles strings for all molecules in a collection, e.g.
>>> print(mols[0:10].smiles())
['CNC(=O)C(C)NC(C)=O', 'O', 'O', 'O', 'O', 'O', 'O', 'O', 'O', 'O']
Note
Note that the smiles string for a water molecule is O
>>> print(mols[0:3].smiles(include_hydrogens=True))
['[H]N(C(=O)C([H])(N([H])C(=O)C([H])([H])[H])C([H])([H])[H])C([H])([H])[H]', '[H]O[H]', '[H]O[H]']
Searching using smiles strings¶
You can search for molecules using smiles strings. To do this,
you need to put smiles before the string, e.g.
>>> mols = sr.load(sr.expand(sr.tutorial_url, "ala.crd", "ala.top"))
>>> print(mols["smiles CNC(=O)C(C)NC(C)=O"])
AtomMatch( size=1
0: [22] CH3:19,N:17,C:15,O:16,CA:9...
)
The result is an sire.mol.AtomMatch object. This is derived from
the atoms container, and it contains all of the atoms that match the
smiles string in the same order in which they appeared in that string.
Sometimes there will be multiple matches for the smiles string, e.g. if there are multiple matches. You can get the number of matches using;
>>> m = mols["smiles CNC(=O)C(C)NC(C)=O"]
>>> print(m.num_groups())
1
and access individual groups using
>>> print(m.group(0))
Selector<SireMol::Atom>( size=22
0: Atom( CH3:19 [ 13.83, 3.94, 18.35] )
1: Atom( N:17 [ 14.97, 4.59, 17.58] )
2: Atom( C:15 [ 15.37, 4.19, 16.43] )
3: Atom( O:16 [ 14.94, 3.17, 15.88] )
4: Atom( CA:9 [ 16.54, 5.03, 15.81] )
...
17: Atom( HB3:14 [ 15.24, 6.18, 14.55] )
18: Atom( H:8 [ 16.68, 3.62, 14.22] )
19: Atom( HH31:1 [ 18.45, 3.49, 12.44] )
20: Atom( HH32:3 [ 20.05, 3.63, 13.29] )
21: Atom( HH33:4 [ 18.80, 2.43, 13.73] )
)
>>> print(m.groups())
[Selector<SireMol::Atom>( size=22
0: Atom( CH3:19 [ 13.83, 3.94, 18.35] )
1: Atom( N:17 [ 14.97, 4.59, 17.58] )
2: Atom( C:15 [ 15.37, 4.19, 16.43] )
3: Atom( O:16 [ 14.94, 3.17, 15.88] )
4: Atom( CA:9 [ 16.54, 5.03, 15.81] )
...
17: Atom( HB3:14 [ 15.24, 6.18, 14.55] )
18: Atom( H:8 [ 16.68, 3.62, 14.22] )
19: Atom( HH31:1 [ 18.45, 3.49, 12.44] )
20: Atom( HH32:3 [ 20.05, 3.63, 13.29] )
21: Atom( HH33:4 [ 18.80, 2.43, 13.73] )
)]
We will discover more about sub-group matching when we try to search using smarts strings.
To simplify searching, you can generate a smiles search string by
passing as_search=True to the smiles()
function. This can be convenient if you are looking to find a molecule
you have already loaded in another collection.
>>> print(mols[0].smiles(as_search=True))
smiles CNC(=O)C(C)NC(C)=O
>>> print(mols[mols[0].smiles(as_search=True)])
AtomMatch( size=1
0: [22] CH3:19,N:17,C:15,O:16,CA:9...
)